使用 Bio.Phylo 進行親緣關係分析
Bio.Phylo 模組在 Biopython 1.54 中引入。它遵循 SeqIO 和 AlignIO 的引導,旨在提供一種獨立於來源資料格式處理親緣關係樹狀圖的通用方法,以及用於 I/O 操作的一致 API。
Bio.Phylo 在一篇開放存取期刊文章 Talevich *et al.* 2012 [Talevich2012] 中有所描述,您可能會覺得它也有幫助。
範例:樹狀圖中有什麼?
為了熟悉該模組,讓我們先從一個已經建構好的樹狀圖開始,並以幾種不同的方式檢查它。然後我們將為分支著色,以使用特殊的 phyloXML 功能,最後將其儲存。
使用您最喜歡的文字編輯器建立一個名為 simple.dnd 的簡單 Newick 檔案,或使用 Biopython 原始碼提供的 simple.dnd
(((A,B),(C,D)),(E,F,G));
此樹狀圖沒有分支長度,只有拓撲結構和標記的末端。(如果您有可用的真實樹狀圖檔案,您可以使用該檔案來跟隨此範例。)
啟動您選擇的 Python 解釋器
$ ipython -pylab
對於互動式工作,使用 -pylab 標誌啟動 IPython 解釋器可以啟用 matplotlib 整合,因此圖形會自動彈出。我們將在此範例期間使用它。
現在,在 Python 中,讀取樹狀圖檔案,並提供檔案名稱和格式名稱。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
將樹狀圖物件以字串形式列印出來,讓我們可以查看整個物件階層。
>>> print(tree)
Tree(rooted=False, weight=1.0)
Clade()
Clade()
Clade()
Clade(name='A')
Clade(name='B')
Clade()
Clade(name='C')
Clade(name='D')
Clade()
Clade(name='E')
Clade(name='F')
Clade(name='G')
Tree 物件包含關於樹狀圖的全域資訊,例如它是有根的還是無根的。它有一個根節點,在該節點下,它是巢狀的節點清單,一直到末端。
函式 draw_ascii 會建立一個簡單的 ASCII 藝術(純文字)樹狀圖。這是一種方便的互動式探索視覺化方法,以防更好的圖形工具不可用。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
>>> Phylo.draw_ascii(tree)
________________________ A
________________________|
| |________________________ B
________________________|
| | ________________________ C
| |________________________|
_| |________________________ D
|
| ________________________ E
| |
|________________________|________________________ F
|
|________________________ G
如果您安裝了 matplotlib 或 pylab,則可以使用 draw 函式建立圖形樹狀圖。
>>> tree.rooted = True
>>> Phylo.draw(tree)
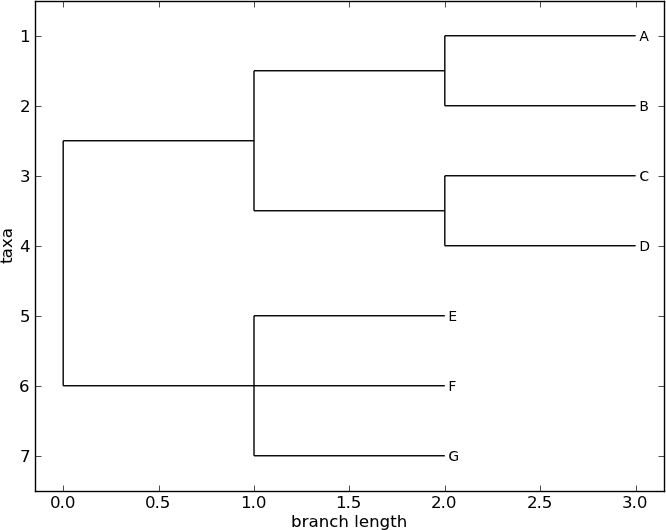
圖 6 使用 Phylo.draw 繪製的有根樹狀圖。
請參閱 圖 6。
為樹狀圖中的分支著色
draw 函式支援在樹狀圖中顯示不同的顏色和分支寬度。從 Biopython 1.59 開始,基本 Clade 物件上提供了 color 和 width 屬性,並且不需要額外的東西即可使用它們。這兩個屬性都指的是通往給定節點的分支,並且會遞迴套用,因此所有後代分支也會在顯示期間繼承分配的寬度和顏色值。
在較早版本的 Biopython 中,這些是 PhyloXML 樹狀圖的特殊功能,並且使用這些屬性需要先將樹狀圖轉換為基本樹狀圖物件的子類別,稱為 Phylogeny,來自 Bio.Phylo.PhyloXML 模組。
在 Biopython 1.55 及更高版本中,這是一種方便的樹狀圖方法
>>> tree = tree.as_phyloxml()
在 Biopython 1.54 中,您可以使用額外的導入來完成相同的事情
>>> from Bio.Phylo.PhyloXML import Phylogeny
>>> tree = Phylogeny.from_tree(tree)
請注意,Newick 和 Nexus 檔案格式不支援分支顏色或寬度,因此如果您在 Bio.Phylo 中使用這些屬性,您將只能以 PhyloXML 格式儲存這些值。(您仍然可以將樹狀圖儲存為 Newick 或 Nexus,但顏色和寬度值將在輸出檔案中跳過。)
現在我們可以開始分配顏色。首先,我們將根節點著色為灰色。我們可以將 24 位元顏色值分配為 RGB 三元組、HTML 樣式的十六進位字串或預定義的顏色名稱來做到這一點。
>>> tree.root.color = (128, 128, 128)
或者
>>> tree.root.color = "#808080"
或者
>>> tree.root.color = "gray"
節點的顏色會被視為向下傳遞到整個節點,因此當我們在此處將根著色時,它會將整個樹狀圖變成灰色。我們可以透過在樹狀圖的較低位置分配不同的顏色來覆寫它。
讓我們以名稱為「E」和「F」的節點的最近共同祖先(MRCA)為目標。common_ancestor 方法會傳回原始樹狀圖中該節點的參考,因此當我們將該節點著色為「鮭魚色」時,該顏色將會顯示在原始樹狀圖中。
>>> mrca = tree.common_ancestor({"name": "E"}, {"name": "F"})
>>> mrca.color = "salmon"
如果我們碰巧知道某個節點在樹狀圖中的確切位置(就巢狀清單項目而言),我們可以透過索引直接跳到樹狀圖中的該位置。在這裡,索引 [0,1] 指的是根的第一個子節點的第二個子節點。
>>> tree.clade[0, 1].color = "blue"
最後,展示我們的成果
>>> Phylo.draw(tree)
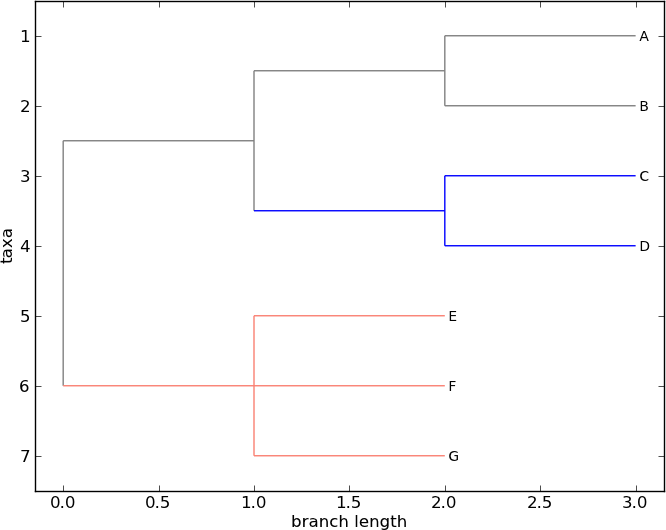
圖 7 使用 Phylo.draw 繪製的著色樹狀圖。
請參閱 圖 7。
請注意,節點的顏色包括通往該節點的分支及其後代。E 和 F 的共同祖先原來就在根的下方,透過這種著色,我們可以清楚地看到樹狀圖的根在哪裡。
我的天啊,我們已經完成了很多!讓我們在這裡休息一下並儲存我們的成果。使用檔案名稱或控制代碼(這裡我們使用標準輸出,以查看將寫入的內容)和格式 phyloxml 呼叫 write 函式。PhyloXML 會儲存我們分配的顏色,因此您可以在其他樹狀圖檢視器(如 Archaeopteryx)中開啟這個 phyloXML 檔案,顏色也會在那裡顯示。
>>> import sys
>>> n = Phylo.write(tree, sys.stdout, "phyloxml")
<phyloxml ...>
<phylogeny rooted="true">
<clade>
<color>
<red>128</red>
<green>128</green>
<blue>128</blue>
</color>
<clade>
<clade>
<clade>
<name>A</name>
</clade>
<clade>
<name>B</name>
</clade>
</clade>
<clade>
<color>
<red>0</red>
<green>0</green>
<blue>255</blue>
</color>
<clade>
<name>C</name>
</clade>
...
</clade>
</phylogeny>
</phyloxml>
>>> n
1
本章的其餘部分更詳細地介紹了 Bio.Phylo 的核心功能。有關使用 Bio.Phylo 的更多範例,請參閱 Biopython.org 上的實作手冊頁面
I/O 函式
與 SeqIO 和 AlignIO 一樣,Phylo 透過四個函式處理檔案輸入和輸出:parse、read、write 和 convert,所有這些函式都支援樹狀圖檔案格式 Newick、NEXUS、phyloXML 和 NeXML,以及比較資料分析本體論 (CDAO)。
read 函式會剖析指定檔案中的單一樹狀圖並傳回它。小心;如果檔案包含多個樹狀圖或沒有樹狀圖,它將引發錯誤。
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Nexus/int_node_labels.nwk", "newick")
>>> print(tree)
Tree(rooted=False, weight=1.0)
Clade(branch_length=75.0, name='gymnosperm')
Clade(branch_length=25.0, name='Coniferales')
Clade(branch_length=25.0)
Clade(branch_length=10.0, name='Tax+nonSci')
Clade(branch_length=90.0, name='Taxaceae')
Clade(branch_length=125.0, name='Cephalotaxus')
...
(範例檔案位於 Biopython 發行版的 Tests/Nexus/ 和 Tests/PhyloXML/ 目錄中。)
若要處理多個(或未知數量的)樹狀圖,請使用 parse 函式,此函式會迭代指定檔案中的每個樹狀圖
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> for tree in trees:
... print(tree)
...
Phylogeny(description='phyloXML allows to use either a "branch_length" attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...', rooted=True)
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
...
使用 write 函式將樹狀圖或可迭代的樹狀圖寫回檔案
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> tree1 = next(trees)
>>> Phylo.write(tree1, "tree1.nwk", "newick")
1
>>> Phylo.write(trees, "other_trees.xml", "phyloxml") # write the remaining trees
12
使用 convert 函式在任何支援的格式之間轉換檔案
>>> Phylo.convert("tree1.nwk", "newick", "tree1.xml", "nexml")
1
>>> Phylo.convert("other_trees.xml", "phyloxml", "other_trees.nex", "nexus")
12
若要使用字串作為輸入或輸出而不是實際檔案,請像使用 SeqIO 和 AlignIO 一樣使用 StringIO
>>> from Bio import Phylo
>>> from io import StringIO
>>> handle = StringIO("(((A,B),(C,D)),(E,F,G));")
>>> tree = Phylo.read(handle, "newick")
檢視和匯出樹狀圖
取得 Tree 物件概觀的最簡單方法是 print 它
>>> from Bio import Phylo
>>> tree = Phylo.read("PhyloXML/example.xml", "phyloxml")
>>> print(tree)
Phylogeny(description='phyloXML allows to use either a "branch_length" attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...', rooted=True)
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
Clade(branch_length=0.23, name='B')
Clade(branch_length=0.4, name='C')
這本質上是 Biopython 用來表示樹狀圖的物件階層的概要。但更有可能的是,您會想要看到樹狀圖的圖形。有三個函式可以執行此操作。
正如我們在範例中所見,draw_ascii 會將樹狀圖的 ascii 藝術繪圖(有根的系統發生圖)列印到標準輸出,如果有的話,也會列印到開啟的檔案控制代碼。並非所有關於樹狀圖的可用資訊都會顯示出來,但它提供了一種在不依賴任何外部依賴項的情況下快速檢視樹狀圖的方法。
>>> tree = Phylo.read("PhyloXML/example.xml", "phyloxml")
>>> Phylo.draw_ascii(tree)
__________________ A
__________|
_| |___________________________________________ B
|
|___________________________________________________________________________ C
draw 函式使用 matplotlib 程式庫繪製更具吸引力的影像。請參閱 API 文件,以瞭解它接受自訂輸出的引數的詳細資訊。
>>> Phylo.draw(tree, branch_labels=lambda c: c.branch_length)
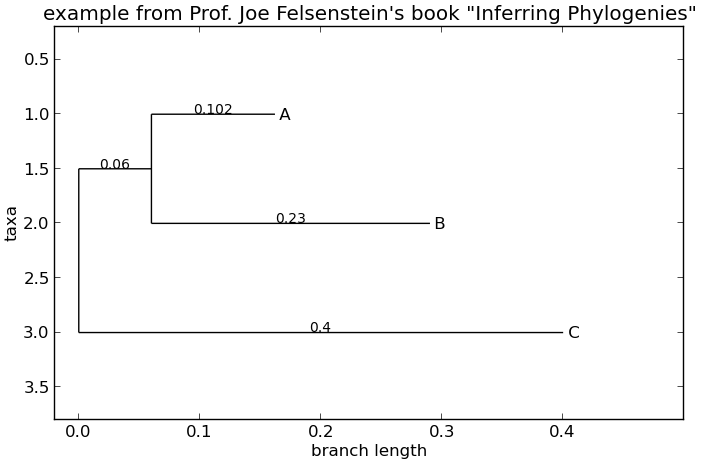
圖 8 使用 draw 函式繪製的簡單有根樹狀圖。
請參閱 圖 8 作為範例。
請參閱 Biopython Wiki 上的 Phylo 頁面 (https://biopython.dev.org.tw/wiki/Phylo),以瞭解 draw_ascii、draw_graphviz 和 to_networkx 中更進階的功能的描述和範例。
使用 Tree 和 Clade 物件
由 parse 和 read 產生的 Tree 物件是遞迴子樹的容器,這些子樹會附加到 Tree 物件的 root 屬性(無論系統發育樹是否實際被視為有根)。Tree 物件具有系統發育的整體資訊,例如是否有根,以及對單一 Clade 的參考;Clade 具有節點和分支群特定的資訊,例如分支長度,以及其自身後代 Clade 實例的列表,這些實例附加在 clades 屬性中。
因此,tree 和 tree.root 之間存在區別。但實際上,您很少需要擔心這一點。為了消除這種差異,Tree 和 Clade 都繼承自 TreeMixin,其中包含常用於搜尋、檢查或修改樹或其任何分支群的方法的實作。這表示 tree 支援的幾乎所有方法,在 tree.root 和其下的任何分支群上也可用。(Clade 也具有 root 屬性,該屬性會返回分支群物件本身。)
搜尋和遍歷方法
為了方便起見,我們提供了一些簡化的方法,可以直接將所有外部或內部節點作為列表返回
get_terminals建立此樹的所有終端(葉)節點的列表。
get_nonterminals建立此樹的所有非終端(內部)節點的列表。
這些方法都包裝了一個可以完全控制樹遍歷的方法 find_clades。另外兩個遍歷方法 find_elements 和 find_any 依賴於相同的核心功能,並接受相同的參數,我們將其稱為「目標規格」,因為沒有更好的描述。這些規格指定在迭代期間樹中的哪些物件將被比對和返回。第一個參數可以是以下任何類型
一個 TreeElement 實例,樹元素將通過身分進行比對 — 因此,使用 Clade 實例作為目標進行搜尋將在樹中找到該分支群;
一個 字串,它會比對樹元素的字串表示 — 特別是,分支群的
name(在 Biopython 1.56 中新增);一個 類別 或 類型,其中相同類型(或子類型)的每個樹元素都將被比對;
一個 字典,其中鍵是樹元素屬性,而值與每個樹元素的對應屬性進行比對。這個會變得更加詳細
如果給定一個
int,它會比對數值相等的屬性,例如 1 將比對 1 或 1.0如果給定一個布林值(True 或 False),則對應的屬性值會被評估為布林值,並檢查是否相同
None比對None如果給定一個字串,則該值將被視為一個正規表示式(它必須比對相應元素屬性中的整個字串,而不僅僅是前綴)。沒有特殊正規表示式字元的給定字串將完全比對字串屬性,因此如果您不使用正規表示式,則不必擔心。例如,在一個分支群名稱為 Foo1、Foo2 和 Foo3 的樹中,
tree.find_clades({"name": "Foo1"})比對 Foo1,{"name": "Foo.*"}比對所有三個分支群,而{"name": "Foo"}不比對任何內容。
由於浮點運算可能會產生一些奇怪的行為,因此我們不支援直接比對
float。相反,請使用布林值True來比對在指定屬性中具有非零值的每個元素,然後使用不等式(或精確數字,如果您喜歡冒險)手動過濾該屬性。如果字典包含多個條目,則比對的元素必須比對每個給定的屬性值 — 請考慮「and」,而不是「or」。
一個 函式,它採用單個參數(它將應用於樹中的每個元素),並返回 True 或 False。為了方便起見,LookupError、AttributeError 和 ValueError 會被抑制,因此這提供了另一種安全的方式來搜尋樹中的浮點值或一些更複雜的特性。
在目標之後,有兩個可選的關鍵字參數
- terminal
— 一個布林值,用於選取或不選取終端分支群(又名葉節點):True 僅搜尋終端分支群,False 搜尋非終端(內部)分支群,而預設值 None 搜尋終端和非終端分支群,以及任何缺少
is_terminal方法的樹元素。- order
— 樹遍歷順序:
"preorder"(預設)是深度優先搜尋,"postorder"是子節點先於父節點的 DFS,而"level"是廣度優先搜尋。
最後,這些方法接受任意關鍵字參數,這些參數的處理方式與字典目標規格相同:鍵表示要搜尋的元素屬性的名稱,而參數值(字串、整數、None 或布林值)會與找到的每個屬性的值進行比較。如果沒有給定關鍵字參數,則會比對任何 TreeElement 類型。此程式碼通常比將字典作為目標規格傳遞更短:tree.find_clades({"name": "Foo1"}) 可以縮短為 tree.find_clades(name="Foo1")。
(在 Biopython 1.56 或更高版本中,這甚至可以更短:tree.find_clades("Foo1"))
現在我們已經掌握了目標規格,以下是用於遍歷樹的方法
find_clades尋找每個包含比對元素的群集。也就是說,使用
find_elements尋找每個元素,但返回相應的分支群物件。(這通常是您想要的。)結果是通過所有比對物件的迭代器,預設情況下進行深度優先搜尋。這不一定與元素在 Newick、Nexus 或 XML 來源檔案中出現的順序相同!
find_elements尋找所有比對給定屬性的樹元素,並返回比對的元素本身。簡單的 Newick 樹沒有複雜的子元素,因此此方法的行為與它們上的
find_clades相同。PhyloXML 樹通常有附加到分支群的複雜物件,因此此方法對於提取這些物件非常有用。find_any返回
find_elements()找到的第一個元素,否則返回 None。這也可用於檢查樹中是否存在任何比對的元素,並且可以在條件中使用。
另外兩個方法有助於在樹中的節點之間導覽
get_path列出樹根(或當前分支群)與給定目標之間直接的分支群。返回沿此路徑的所有分支群物件的列表,以給定目標結束,但不包括根分支群。
trace列出此樹中兩個目標之間的所有分支群物件。不包括開始,包括結束。
資訊方法
這些方法提供有關整棵樹(或任何分支群)的資訊。
common_ancestor尋找所有給定目標的最近共同祖先。(這將是一個 Clade 物件)。如果沒有給定目標,則返回當前分支群的根(調用此方法的分支群);如果給定 1 個目標,則返回目標本身。但是,如果在當前樹(或分支群)中找不到任何指定的目標,則會引發例外狀況。
count_terminals計算樹中終端(葉)節點的數量。
depths建立樹分支群到深度的對應關係。結果是一個字典,其中鍵是樹中的所有 Clade 實例,值是從根到每個分支群的距離(包括終端)。預設情況下,距離是通往分支群的累積分支長度,但使用
unit_branch_lengths=True選項,只會計算分支的數量(樹中的層級)。distance計算兩個目標之間的分支長度總和。如果僅指定一個目標,則另一個目標是此樹的根。
total_branch_length計算此樹中所有分支長度的總和。這通常在系統發育學中簡稱為樹的「長度」,但我們使用更明確的名稱,以避免與 Python 術語混淆。
其餘這些方法是布林檢查
is_bifurcating如果樹是嚴格二叉的,則為 True;即所有節點都有 2 個或 0 個子節點(分別為內部或外部)。根可能具有 3 個後代,並且仍然被視為二叉樹的一部分。
is_monophyletic測試所有給定目標是否構成完整的分支群 — 即,是否存在一個分支群,使其終端與給定目標的集合相同。目標應該是樹的終端。為方便起見,如果目標是單系群,則此方法返回目標的共同祖先(MCRA)(而不是值
True),否則返回False。is_parent_of如果目標是此樹的後代,則為 True — 不一定要是直接後代。要檢查一個演化支的直接後代,只需使用列表成員測試:
if subclade in clade: ...is_preterminal如果所有直接後代都是終端節點則為 True;如果任何直接後代不是終端節點則為 False。
修改方法
這些方法會就地修改樹狀結構。如果您想保留原始樹狀結構的完整性,請先使用 Python 的 copy 模組完整複製樹狀結構。
tree = Phylo.read("example.xml", "phyloxml")
import copy
newtree = copy.deepcopy(tree)
collapse從樹狀結構中刪除目標,將其子節點重新連結到其父節點。
collapse_all摺疊此樹的所有後代,僅留下終端節點。分支長度會被保留,即到每個終端節點的距離保持不變。使用目標規格(見上文),僅摺疊符合規格的內部節點。
ladderize根據終端節點的數量就地排序演化支。預設情況下,最深的演化支會放在最後。使用
reverse=True將演化支從最深到最淺排序。prune從樹狀結構中修剪終端演化支。如果分類單元來自分叉,則連接節點將被摺疊,其分支長度將添加到剩餘的終端節點。這可能不再是一個有意義的值。
root_with_outgroup使用包含給定目標的外群演化支,即外群的共同祖先,重新定位此樹的根。此方法僅適用於樹物件,不適用於演化支。
如果外群與 self.root 相同,則不會發生任何變更。如果外群演化支是終端節點(例如,給定的外群是單一終端節點),則會建立一個新的分叉根演化支,並將長度為 0 的分支連結到給定的外群。否則,外群底部的內部節點會成為整個樹的三叉根。如果原始根是分叉的,則會從樹中刪除。
在所有情況下,樹的總分支長度保持不變。
root_at_midpoint在此樹的兩個最遠頂端之間計算的中點重新定位此樹的根。(這在底層使用了
root_with_outgroup。)split產生 n 個(預設為 2 個)新的後代。在物種樹中,這是一個物種形成事件。新的演化支具有給定的
branch_length,並且與此演化支的根相同的名稱加上整數後綴(從 0 開始計數)— 例如,分割名為「A」的演化支會產生子演化支「A0」和「A1」。
請參閱 Biopython 維基上的 Phylo 頁面(https://biopython.dev.org.tw/wiki/Phylo)以獲取有關使用可用方法的更多範例。
PhyloXML 樹的功能
phyloXML 檔案格式包含用於使用其他資料類型和視覺提示來註解樹狀結構的欄位。
請參閱 Biopython 維基上的 PhyloXML 頁面(https://biopython.dev.org.tw/wiki/PhyloXML)以取得有關使用 PhyloXML 提供的其他註解功能的描述和範例。
執行外部應用程式
雖然 Bio.Phylo 本身不會從序列比對中推斷樹狀結構,但有可用的第三方程式可以執行此操作。可以使用 subprocess 模組從 Python 內部存取這些程式。
以下是一個範例,說明如何使用 Python 指令碼與 PhyML 互動(http://www.atgc-montpellier.fr/phyml/)。該程式接受 phylip-relaxed 格式(即 Phylip 格式,但對分類單元名稱沒有 10 個字元的限制)的輸入序列比對以及各種選項。
>>> import subprocess
>>> cmd = "phyml -i Tests/Phylip/random.phy"
>>> results = subprocess.run(cmd, shell=True, stdout=subprocess.PIPE, text=True)
「stdout = subprocess.PIPE」引數使程式的輸出可透過「results.stdout」進行偵錯(「stderr」也可以執行相同操作),而「text=True」使傳回的資訊成為 Python 字串,而不是「bytes」物件。
這會產生一個樹狀結構檔案和一個統計資料檔案,名稱為 [輸入檔案名稱]_phyml_tree.txt 和 [輸入檔案名稱]_phyml_stats.txt。樹狀結構檔案採用 Newick 格式
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Phylip/random.phy_phyml_tree.txt", "newick")
>>> Phylo.draw_ascii(tree)
__________________ F
|
| I
|
_| ________ C
| ________|
| | | , J
| | |________|
| | | , H
|___________| |__________|
| |______________ D
|
, G
|
| , E
|________________|
| ___________________________ A
|________________|
|_________ B
subprocess 模組還可以用於與任何其他提供命令列介面的程式互動,例如 RAxML(https://sco.h-its.org/exelixis/software.html)、FastTree(http://www.microbesonline.org/fasttree/)、dnaml 和 protml。
PAML 整合
Biopython 1.58 為 PAML 提供了支援(http://abacus.gene.ucl.ac.uk/software/paml.html),這是一套透過最大概似法進行系統發生分析的程式。目前已實作程式 codeml、baseml 和 yn00。由於 PAML 使用控制檔案而不是命令列引數來控制執行階段選項,因此此封裝器的使用方式與 Biopython 中其他應用程式封裝器的格式不同。
典型的使用流程是初始化 PAML 物件,指定序列比對檔案、樹狀結構檔案、輸出檔案和工作目錄。接下來,透過 set_options() 方法或讀取現有的控制檔案來設定執行階段選項。最後,透過 run() 方法執行程式,並將輸出檔案自動剖析為結果字典。
以下是 codeml 的典型用法範例
>>> from Bio.Phylo.PAML import codeml
>>> cml = codeml.Codeml()
>>> cml.alignment = "Tests/PAML/Alignments/alignment.phylip"
>>> cml.tree = "Tests/PAML/Trees/species.tree"
>>> cml.out_file = "results.out"
>>> cml.working_dir = "./scratch"
>>> cml.set_options(
... seqtype=1,
... verbose=0,
... noisy=0,
... RateAncestor=0,
... model=0,
... NSsites=[0, 1, 2],
... CodonFreq=2,
... cleandata=1,
... fix_alpha=1,
... kappa=4.54006,
... )
>>> results = cml.run()
>>> ns_sites = results.get("NSsites")
>>> m0 = ns_sites.get(0)
>>> m0_params = m0.get("parameters")
>>> print(m0_params.get("omega"))
也可以使用模組的 read() 函式來剖析現有的輸出檔案
>>> results = codeml.read("Tests/PAML/Results/codeml/codeml_NSsites_all.out")
>>> print(results.get("lnL max"))
此新模組的詳細文件目前位於 Biopython 維基上:https://biopython.dev.org.tw/wiki/PAML
未來計畫
Bio.Phylo 正在積極開發中。以下是我們可能會在未來版本中新增的一些功能
- 新方法
通常用於操作樹狀結構或演化支物件的實用函式會先出現在 Biopython 維基上,以便一般使用者可以測試它們並判斷它們是否有用,然後我們才會將它們新增到 Bio.Phylo 中
- Bio.Nexus 連接埠
此模組的大部分是在 2009 年 Google Summer of Code 期間,在 NESCent 的贊助下編寫的,作為一個實作 phyloXML 資料格式的 Python 支援專案(請參閱 PhyloXML 樹的功能)。透過將現有 Bio.Nexus 模組的一部分移植到 Bio.Phylo 使用的新類別,新增了對 Newick 和 Nexus 格式的支援。
目前,Bio.Nexus 包含一些尚未移植到 Bio.Phylo 類別的實用功能 — 尤其是計算一致樹。如果您發現 Bio.Phylo 中缺少某些功能,請嘗試檢查 Bio.Nexus 中是否存在該功能。
我們樂於接受任何關於改進此模組功能和可用性的建議;只需在郵件清單或我們的錯誤資料庫中通知我們即可。
最後,如果您需要 Phylo 模組中尚未包含的其他功能,請檢查在其他高品質的系統發生學 Python 程式庫(例如 DendroPy (https://dendropy.org/) 或 PyCogent (http://pycogent.org/))中是否可用。由於這些程式庫也支援系統發生樹的標準檔案格式,您可以輕鬆地透過寫入暫存檔案或 StringIO 物件在程式庫之間傳輸資料。