食譜 – 使用 Biopython 的酷炫方法
Biopython 現在有兩個「食譜」範例集合 – 本章(已納入本教學多年並逐漸成長),以及 https://biopython.dev.org.tw/wiki/Category:Cookbook,這是使用者在我們的 wiki 上貢獻的集合。
我們正在努力鼓勵 Biopython 使用者將他們自己的範例貢獻到 wiki 上。除了幫助社群之外,分享類似範例的一個直接好處是,您還可以從其他 Biopython 使用者和開發人員那裡獲得程式碼的回饋 – 這可以幫助您改進所有的 Python 程式碼。
從長遠來看,我們可能會將本章中的所有範例移到 wiki 中,或移到教學的其他位置。
處理序列檔案
本節顯示更多序列輸入/輸出的範例,使用第 序列輸入/輸出 章中描述的 Bio.SeqIO 模組。
篩選序列檔案
通常,您會有一個包含許多序列的大型檔案(例如,基因的 FASTA 檔案,或讀取的 FASTQ 或 SFF 檔案),一個單獨的較短列表,其中包含感興趣的序列子集的 ID,並希望為此子集建立一個新的序列檔案。
假設 ID 列表位於一個簡單的文字檔中,作為每行的第一個字詞。這可能是一個表格檔案,其中第一欄是 ID。嘗試以下方法
from Bio import SeqIO
input_file = "big_file.sff"
id_file = "short_list.txt"
output_file = "short_list.sff"
with open(id_file) as id_handle:
wanted = set(line.rstrip("\n").split(None, 1)[0] for line in id_handle)
print("Found %i unique identifiers in %s" % (len(wanted), id_file))
records = (r for r in SeqIO.parse(input_file, "sff") if r.id in wanted)
count = SeqIO.write(records, output_file, "sff")
print("Saved %i records from %s to %s" % (count, input_file, output_file))
if count < len(wanted):
print("Warning %i IDs not found in %s" % (len(wanted) - count, input_file))
請注意,我們使用 Python set 而不是 list,這使得測試成員資格更快。
如第 低階 FASTA 和 FASTQ 解析器 節中所討論,對於大型 FASTA 或 FASTQ 檔案,為了速度起見,您最好不要使用高階 SeqIO 介面,而是直接使用字串。下一個範例示範如何使用 FASTQ 檔案執行此操作 – 這會更複雜
from Bio.SeqIO.QualityIO import FastqGeneralIterator
input_file = "big_file.fastq"
id_file = "short_list.txt"
output_file = "short_list.fastq"
with open(id_file) as id_handle:
# Taking first word on each line as an identifier
wanted = set(line.rstrip("\n").split(None, 1)[0] for line in id_handle)
print("Found %i unique identifiers in %s" % (len(wanted), id_file))
with open(input_file) as in_handle:
with open(output_file, "w") as out_handle:
for title, seq, qual in FastqGeneralIterator(in_handle):
# The ID is the first word in the title line (after the @ sign):
if title.split(None, 1)[0] in wanted:
# This produces a standard 4-line FASTQ entry:
out_handle.write("@%s\n%s\n+\n%s\n" % (title, seq, qual))
count += 1
print("Saved %i records from %s to %s" % (count, input_file, output_file))
if count < len(wanted):
print("Warning %i IDs not found in %s" % (len(wanted) - count, input_file))
產生隨機化的基因組
假設您正在查看基因組序列,尋找一些序列特徵 – 可能是極端的局部 GC% 偏差,或可能的限制酶切位點。一旦您的 Python 程式碼可以在真實基因組上運作,對相同基因組的隨機版本執行相同的搜尋進行統計分析可能是有意義的(畢竟,您找到的任何「特徵」都可能只是偶然存在)。
對於本討論,我們將使用來自鼠疫耶爾森菌微型變種的 pPCP1 質體的 GenBank 檔案。該檔案包含在 Biopython 單位測試的 GenBank 資料夾下,或者您可以從我們的網站 NC_005816.gb 取得。此檔案包含一個且僅一個記錄,因此我們可以使用 Bio.SeqIO.read() 函數將其讀入為 SeqRecord
>>> from Bio import SeqIO
>>> original_rec = SeqIO.read("NC_005816.gb", "genbank")
那麼,我們如何產生原始序列的隨機排序版本?我會使用內建的 Python random 模組來實現,特別是函數 random.shuffle – 但這適用於 Python 列表。我們的序列是 Seq 物件,因此為了對其進行排序,我們需要將其轉換為列表
>>> import random
>>> nuc_list = list(original_rec.seq)
>>> random.shuffle(nuc_list) # acts in situ!
現在,為了使用 Bio.SeqIO 輸出排序後的序列,我們需要使用此排序後的列表建構一個新的 SeqRecord,其中包含一個新的 Seq 物件。為了做到這一點,我們需要將核苷酸列表(單字母字串)轉換為長字串 – 標準的 Python 方法是使用字串物件的 join 方法。
>>> from Bio.Seq import Seq
>>> from Bio.SeqRecord import SeqRecord
>>> shuffled_rec = SeqRecord(
... Seq("".join(nuc_list)), id="Shuffled", description="Based on %s" % original_rec.id
... )
讓我們將所有這些片段放在一起,建立一個完整的 Python 腳本,該腳本會產生一個包含 30 個原始序列隨機排序版本的 FASTA 檔案。
第一個版本只使用一個大的 for 迴圈,並將記錄逐一寫出(使用第 以格式化字串取得您的 SeqRecord 物件 節中描述的 SeqRecord 的 format 方法)
import random
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio import SeqIO
original_rec = SeqIO.read("NC_005816.gb", "genbank")
with open("shuffled.fasta", "w") as output_handle:
for i in range(30):
nuc_list = list(original_rec.seq)
random.shuffle(nuc_list)
shuffled_rec = SeqRecord(
Seq("".join(nuc_list)),
id="Shuffled%i" % (i + 1),
description="Based on %s" % original_rec.id,
)
output_handle.write(shuffled_rec.format("fasta"))
我個人比較喜歡以下版本,它使用一個函數來排序記錄,並使用產生器運算式而不是 for 迴圈
import random
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio import SeqIO
def make_shuffle_record(record, new_id):
nuc_list = list(record.seq)
random.shuffle(nuc_list)
return SeqRecord(
Seq("".join(nuc_list)),
id=new_id,
description="Based on %s" % original_rec.id,
)
original_rec = SeqIO.read("NC_005816.gb", "genbank")
shuffled_recs = (
make_shuffle_record(original_rec, "Shuffled%i" % (i + 1)) for i in range(30)
)
SeqIO.write(shuffled_recs, "shuffled.fasta", "fasta")
翻譯 CDS 條目的 FASTA 檔案
假設您有一些生物的 CDS 條目的輸入檔案,並且您想要產生一個包含其蛋白質序列的新 FASTA 檔案。也就是說,從原始檔案中取得每個核苷酸序列,並進行翻譯。在第 翻譯 節中,我們看到了如何使用 Seq 物件的 translate 方法,以及可讓您正確翻譯替代起始密碼子的選用 cds 引數。
我們可以將此與 Bio.SeqIO 結合使用,如第 將序列檔案轉換為其反向互補序列 節中的反向互補範例所示。重點是,對於每個核苷酸 SeqRecord,我們需要建立一個蛋白質 SeqRecord – 並處理其命名。
您可以編寫自己的函數來執行此操作,為您的序列選擇合適的蛋白質識別符,以及適當的遺傳密碼。在此範例中,我們只使用預設表格並在識別符中新增前綴
from Bio.SeqRecord import SeqRecord
def make_protein_record(nuc_record):
"""Returns a new SeqRecord with the translated sequence (default table)."""
return SeqRecord(
seq=nuc_record.seq.translate(cds=True),
id="trans_" + nuc_record.id,
description="translation of CDS, using default table",
)
然後,我們可以使用此函數將輸入的核苷酸記錄轉換為可供輸出的蛋白質記錄。一種優雅且記憶體效率高的方法是使用產生器運算式
from Bio import SeqIO
proteins = (
make_protein_record(nuc_rec)
for nuc_rec in SeqIO.parse("coding_sequences.fasta", "fasta")
)
SeqIO.write(proteins, "translations.fasta", "fasta")
這應該適用於任何完整的編碼序列的 FASTA 檔案。如果您正在使用部分編碼序列,您可能更喜歡在上面的範例中使用 nuc_record.seq.translate(to_stop=True),因為這不會檢查有效的起始密碼子等。
將 FASTA 檔案中的序列轉為大寫
您通常會從合作者那裡收到 FASTA 檔案形式的資料,有時序列可能混合使用大小寫字母。在某些情況下,這是刻意的(例如,小寫字母表示品質較差的區域),但通常並不重要。您可能想要編輯檔案以使所有內容保持一致(例如,全部大寫),並且您可以使用 SeqRecord 物件的 upper() 方法輕鬆完成此操作(在 Biopython 1.55 中新增)
from Bio import SeqIO
records = (rec.upper() for rec in SeqIO.parse("mixed.fas", "fasta"))
count = SeqIO.write(records, "upper.fas", "fasta")
print("Converted %i records to upper case" % count)
這是如何運作的?第一行只是匯入 Bio.SeqIO 模組。第二行是有趣的部分 – 這是一個 Python 產生器運算式,它會提供從輸入檔案 (mixed.fas) 解析的每個記錄的大寫版本。在第三行中,我們將此產生器運算式提供給 Bio.SeqIO.write() 函數,它會將新的大寫記錄儲存到我們的輸出檔案 (upper.fas)。
我們使用產生器運算式(而不是列表或列表推導式)的原因是,這表示一次只將一個記錄保存在記憶體中。如果您正在處理數百萬條條目的大型檔案,這可能非常重要。
排序序列檔案
假設您想要按長度排序序列檔案(例如,來自組合的一組 contigs),並且您正在使用 Bio.SeqIO 可以讀取、寫入(和索引)的 FASTA 或 FASTQ 之類的檔案格式。
如果檔案夠小,您可以一次將其全部載入記憶體中,作為 SeqRecord 物件的列表,對列表進行排序,然後儲存
from Bio import SeqIO
records = list(SeqIO.parse("ls_orchid.fasta", "fasta"))
records.sort(key=lambda r: len(r))
SeqIO.write(records, "sorted_orchids.fasta", "fasta")
唯一巧妙的地方是指定一個比較方法,用於如何排序記錄(在這裡我們按長度排序它們)。如果您想要先顯示最長的記錄,您可以翻轉比較或使用 reverse 引數
from Bio import SeqIO
records = list(SeqIO.parse("ls_orchid.fasta", "fasta"))
records.sort(key=lambda r: -len(r))
SeqIO.write(records, "sorted_orchids.fasta", "fasta")
現在這非常簡單 – 但是如果您有一個非常大的檔案並且無法像這樣將其全部載入記憶體中,會發生什麼情況?例如,您可能有一些下一代定序讀取需要按長度排序。這可以使用 Bio.SeqIO.index() 函數來解決。
from Bio import SeqIO
# Get the lengths and ids, and sort on length
len_and_ids = sorted(
(len(rec), rec.id) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")
)
ids = reversed([id for (length, id) in len_and_ids])
del len_and_ids # free this memory
record_index = SeqIO.index("ls_orchid.fasta", "fasta")
records = (record_index[id] for id in ids)
SeqIO.write(records, "sorted.fasta", "fasta")
首先,我們使用 Bio.SeqIO.parse() 掃描檔案一次,將記錄的識別碼及其長度記錄在一個元組列表中。然後,我們對這個列表進行排序,使其按照長度順序排列,並捨棄長度。使用這個已排序的識別碼列表,Bio.SeqIO.index() 允許我們逐一檢索記錄,並將它們傳遞給 Bio.SeqIO.write() 進行輸出。
這些範例都使用 Bio.SeqIO 將記錄解析為 SeqRecord 物件,然後使用 Bio.SeqIO.write() 輸出。如果想要排序 Bio.SeqIO.write() 不支援的檔案格式(例如純文字 SwissProt 格式)該怎麼辦?以下是另一個解決方案,它使用 Biopython 1.54 中添加到 Bio.SeqIO.index() 的 get_raw() 方法(請參閱 取得記錄的原始資料)。
from Bio import SeqIO
# Get the lengths and ids, and sort on length
len_and_ids = sorted(
(len(rec), rec.id) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")
)
ids = reversed([id for (length, id) in len_and_ids])
del len_and_ids # free this memory
record_index = SeqIO.index("ls_orchid.fasta", "fasta")
with open("sorted.fasta", "wb") as out_handle:
for id in ids:
out_handle.write(record_index.get_raw(id))
請注意,從 Python 3 開始,我們必須以二進制模式開啟檔案進行寫入,因為 get_raw() 方法會傳回 bytes 物件。
額外的好處是,因為它不會第二次將資料解析為 SeqRecord 物件,所以速度應該會更快。如果只想將此方法用於 FASTA 格式,我們可以透過使用低階 FASTA 解析器來取得記錄識別碼和長度,進一步加快速度。
from Bio.SeqIO.FastaIO import SimpleFastaParser
from Bio import SeqIO
# Get the lengths and ids, and sort on length
with open("ls_orchid.fasta") as in_handle:
len_and_ids = sorted(
(len(seq), title.split(None, 1)[0])
for title, seq in SimpleFastaParser(in_handle)
)
ids = reversed([id for (length, id) in len_and_ids])
del len_and_ids # free this memory
record_index = SeqIO.index("ls_orchid.fasta", "fasta")
with open("sorted.fasta", "wb") as out_handle:
for id in ids:
out_handle.write(record_index.get_raw(id))
FASTQ 檔案的簡單品質過濾
FASTQ 檔案格式是在 Sanger 引入的,現在廣泛用於保存核苷酸序列讀取資料及其品質分數。FASTQ 檔案(以及相關的 QUAL 檔案)是每個字母註解的絕佳範例,因為序列中的每個核苷酸都有一個相關的品質分數。任何每個字母的註解都會以列表、元組或字串的形式,保存在 SeqRecord 的 letter_annotations 字典中(元素數量與序列長度相同)。
常見的任務是取得大量的序列讀取資料,並根據其品質分數進行過濾(或裁剪)。以下範例非常簡單,但應說明如何使用 SeqRecord 物件中的品質資料的基本知識。我們在這裡要做的只是讀入 FASTQ 資料檔案,並過濾它,只選取 PHRED 品質分數都高於某個閾值(這裡為 20)的記錄。
在這個範例中,我們將使用從 ENA 序列讀取檔案庫下載的一些真實資料:ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz (2MB),解壓縮後會產生一個 19MB 的檔案 SRR020192.fastq。這是一些來自感染病毒的加州海獅的 Roche 454 GS FLX 單端資料(詳情請參閱 https://www.ebi.ac.uk/ena/data/view/SRS004476)。
首先,讓我們計算讀取次數
from Bio import SeqIO
count = 0
for rec in SeqIO.parse("SRR020192.fastq", "fastq"):
count += 1
print("%i reads" % count)
現在,讓我們執行一個簡單的過濾,以達到 PHRED 品質至少為 20 的要求
from Bio import SeqIO
good_reads = (
rec
for rec in SeqIO.parse("SRR020192.fastq", "fastq")
if min(rec.letter_annotations["phred_quality"]) >= 20
)
count = SeqIO.write(good_reads, "good_quality.fastq", "fastq")
print("Saved %i reads" % count)
這從 \(41892\) 個讀取資料中提取了 \(14580\) 個讀取資料。更明智的做法是修剪品質讀取資料,但這僅作為範例。
FASTQ 檔案可能包含數百萬個條目,因此最好避免一次將它們全部載入記憶體中。此範例使用產生器表達式,這表示一次只會建立一個 SeqRecord,從而避免任何記憶體限制。
請注意,這裡使用低階 FastqGeneralIterator 解析器會更快(請參閱 低階 FASTA 和 FASTQ 解析器),但這不會將品質字串轉換為整數分數。
修剪引物序列
在這個範例中,我們將假設 GATGACGGTGT 是我們要在某些 FASTQ 格式的讀取資料中尋找的 5' 引物序列。如同以上範例,我們將使用從 ENA 下載的 SRR020192.fastq 檔案 (ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz)。
藉由使用主要的 Bio.SeqIO 介面,相同的方法也適用於任何其他支援的檔案格式(例如 FASTA 檔案)。不過,對於大型 FASTQ 檔案,使用低階 FastqGeneralIterator 解析器會更快(請參閱先前的範例以及 低階 FASTA 和 FASTQ 解析器)。
此程式碼將 Bio.SeqIO 與產生器表達式搭配使用(以避免一次將所有序列載入記憶體),並使用 Seq 物件的 startswith 方法來查看讀取資料是否以引物序列開始
from Bio import SeqIO
primer_reads = (
rec
for rec in SeqIO.parse("SRR020192.fastq", "fastq")
if rec.seq.startswith("GATGACGGTGT")
)
count = SeqIO.write(primer_reads, "with_primer.fastq", "fastq")
print("Saved %i reads" % count)
這應該可以從 SRR014849.fastq 中找到 \(13819\) 個讀取資料,並將其儲存到新的 FASTQ 檔案 with_primer.fastq。
現在,假設您想要建立一個 FASTQ 檔案,其中包含這些讀取資料,但已移除引物序列?這只是一個小小的變更,因為我們可以切片 SeqRecord(請參閱 切片 SeqRecord)以移除前十一個字母(我們的引物長度)
from Bio import SeqIO
trimmed_primer_reads = (
rec[11:]
for rec in SeqIO.parse("SRR020192.fastq", "fastq")
if rec.seq.startswith("GATGACGGTGT")
)
count = SeqIO.write(trimmed_primer_reads, "with_primer_trimmed.fastq", "fastq")
print("Saved %i reads" % count)
同樣地,這應該可以從 SRR020192.fastq 中提取 \(13819\) 個讀取資料,但這次會去掉前十個字元,並將其儲存到另一個新的 FASTQ 檔案 with_primer_trimmed.fastq。
現在,假設您想要建立一個新的 FASTQ 檔案,其中這些讀取資料已移除引物,但所有其他讀取資料都保持原樣?如果我們仍想使用產生器表達式,那麼定義我們自己的修剪函式可能最清楚
from Bio import SeqIO
def trim_primer(record, primer):
if record.seq.startswith(primer):
return record[len(primer) :]
else:
return record
trimmed_reads = (
trim_primer(record, "GATGACGGTGT")
for record in SeqIO.parse("SRR020192.fastq", "fastq")
)
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print("Saved %i reads" % count)
這需要更長的時間,因為這次輸出檔案包含所有 \(41892\) 個讀取資料。同樣地,我們使用了產生器表達式來避免任何記憶體問題。您也可以使用產生器函式,而不是產生器表達式。
from Bio import SeqIO
def trim_primers(records, primer):
"""Removes perfect primer sequences at start of reads.
This is a generator function, the records argument should
be a list or iterator returning SeqRecord objects.
"""
len_primer = len(primer) # cache this for later
for record in records:
if record.seq.startswith(primer):
yield record[len_primer:]
else:
yield record
original_reads = SeqIO.parse("SRR020192.fastq", "fastq")
trimmed_reads = trim_primers(original_reads, "GATGACGGTGT")
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print("Saved %i reads" % count)
如果您想要執行更複雜的操作,其中只保留某些記錄,則此形式更具彈性,如下一個範例所示。
修剪接合子序列
這基本上是先前範例的簡單延伸。我們將假設 GATGACGGTGT 是某些 FASTQ 格式讀取資料中的接合子序列,再次使用來自 NCBI 的 SRR020192.fastq 檔案 (ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz)。
不過,這次我們將在讀取資料中的任何位置尋找該序列,而不僅僅是在最開始
from Bio import SeqIO
def trim_adaptors(records, adaptor):
"""Trims perfect adaptor sequences.
This is a generator function, the records argument should
be a list or iterator returning SeqRecord objects.
"""
len_adaptor = len(adaptor) # cache this for later
for record in records:
index = record.seq.find(adaptor)
if index == -1:
# adaptor not found, so won't trim
yield record
else:
# trim off the adaptor
yield record[index + len_adaptor :]
original_reads = SeqIO.parse("SRR020192.fastq", "fastq")
trimmed_reads = trim_adaptors(original_reads, "GATGACGGTGT")
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print("Saved %i reads" % count)
因為在此範例中我們使用 FASTQ 輸入檔案,所以 SeqRecord 物件具有品質分數的每個字母註解。透過切片 SeqRecord 物件,修剪後的記錄會使用適當的分數,因此我們也可以將其輸出為 FASTQ 檔案。
與先前範例的輸出相比,我們只尋找每個讀取資料開頭的引物/接合子,您可能會發現某些修剪後的讀取資料在修剪後會非常短(例如,如果在中間而不是在開頭附近找到接合子)。因此,讓我們也加入最低長度要求
from Bio import SeqIO
def trim_adaptors(records, adaptor, min_len):
"""Trims perfect adaptor sequences, checks read length.
This is a generator function, the records argument should
be a list or iterator returning SeqRecord objects.
"""
len_adaptor = len(adaptor) # cache this for later
for record in records:
len_record = len(record) # cache this for later
if len(record) < min_len:
# Too short to keep
continue
index = record.seq.find(adaptor)
if index == -1:
# adaptor not found, so won't trim
yield record
elif len_record - index - len_adaptor >= min_len:
# after trimming this will still be long enough
yield record[index + len_adaptor :]
original_reads = SeqIO.parse("SRR020192.fastq", "fastq")
trimmed_reads = trim_adaptors(original_reads, "GATGACGGTGT", 100)
count = SeqIO.write(trimmed_reads, "trimmed.fastq", "fastq")
print("Saved %i reads" % count)
藉由變更格式名稱,您可以將此方法應用於 FASTA 檔案。此程式碼也可以延伸為執行模糊比對,而不是精確比對(或許使用成對比對,或將讀取品質分數納入考量),但速度會慢很多。
轉換 FASTQ 檔案
在 在序列檔案格式之間轉換 中,我們示範了如何使用 Bio.SeqIO 在兩種檔案格式之間進行轉換。在這裡,我們將更詳細地介紹第二代 DNA 定序中使用的 FASTQ 檔案。請參考 Cock et al. (2010) [Cock2010] 以取得更詳細的說明。FASTQ 檔案會將 DNA 序列(作為字串)和相關的讀取品質儲存起來。
PHRED 分數(用於大多數 FASTQ 檔案,以及 QUAL 檔案、ACE 檔案和 SFF 檔案)已成為表示定序錯誤機率(此處以 \(P_e\) 表示)的事實標準,方法是使用簡單的以 10 為底的對數轉換
這表示錯誤的讀取 (\(P_e = 1\)) 會獲得 PHRED 品質 \(0\),而像 \(P_e = 0.00001\) 這種非常好的讀取會獲得 PHRED 品質 \(50\)。雖然原始定序資料的品質高於此數值的情況很少見,但經過讀取資料對應或組裝等後處理後,可能會出現高達 \(90\) 左右的品質(實際上,MAQ 工具允許 PHRED 分數在 0 到 93(含)的範圍內)。
FASTQ 格式有可能成為在單個純文字檔案中儲存定序讀取字母和品質分數的事實標準。唯一的美中不足是,至少有三個版本的 FASTQ 格式不相容且難以區分…
原始的 Sanger FASTQ 格式使用 PHRED 品質評分,並以 ASCII 偏移量 33 編碼。NCBI 在其短序列讀取檔案庫中使用此格式。我們在
Bio.SeqIO中稱此格式為fastq(或fastq-sanger)。Solexa (後來被 Illumina 收購) 引入了他們自己的版本,使用 Solexa 品質評分,並以 ASCII 偏移量 64 編碼。我們稱此格式為
fastq-solexa。Illumina 管線 1.3 版之後產生的 FASTQ 檔案使用 PHRED 品質評分(更一致),但以 ASCII 偏移量 64 編碼。我們稱此格式為
fastq-illumina。
Solexa 品質評分使用不同的對數轉換來定義
鑑於 Solexa/Illumina 現在已在其管線 1.3 版中改為使用 PHRED 評分,Solexa 品質評分將逐漸不再使用。如果您將誤差估計值 (\(P_e\)) 等同,則這兩個方程式可允許在兩個評分系統之間進行轉換 - Biopython 在 Bio.SeqIO.QualityIO 模組中包含執行此操作的函數,如果您使用 Bio.SeqIO 將舊的 Solexa/Illumina 檔案轉換為標準的 Sanger FASTQ 檔案,就會呼叫這些函數
from Bio import SeqIO
SeqIO.convert("solexa.fastq", "fastq-solexa", "standard.fastq", "fastq")
如果您想要轉換新的 Illumina 1.3+ FASTQ 檔案,唯一會更改的是 ASCII 偏移量,因為雖然編碼方式不同,但評分都是 PHRED 品質評分
from Bio import SeqIO
SeqIO.convert("illumina.fastq", "fastq-illumina", "standard.fastq", "fastq")
請注意,像這樣使用 Bio.SeqIO.convert() 會比結合使用 Bio.SeqIO.parse() 和 Bio.SeqIO.write() 快很多,因為優化的程式碼用於在 FASTQ 變體之間轉換(也用於 FASTQ 到 FASTA 的轉換)。
對於高品質的讀取資料,PHRED 和 Solexa 評分大致相等,這表示由於 fasta-solexa 和 fastq-illumina 格式都使用 ASCII 偏移量 64,因此檔案幾乎相同。這是 Illumina 的刻意設計選擇,表示預期使用舊 fasta-solexa 樣式檔案的應用程式,使用較新的 fastq-illumina 檔案(在良好資料下)可能也沒問題。當然,這兩個變體都與 Sanger、NCBI 和其他地方使用的原始 FASTQ 標準非常不同(格式名稱為 fastq 或 fastq-sanger)。
如需更多詳細資訊,請參閱內建說明(也可在 Bio.SeqIO.QualityIO 中找到)
>>> from Bio.SeqIO import QualityIO
>>> help(QualityIO)
將 FASTA 和 QUAL 檔案轉換為 FASTQ 檔案
FASTQ 檔案保存序列和其品質字串。FASTA 檔案僅保存序列,而 QUAL 檔案僅保存品質。因此,單一 FASTQ 檔案可以轉換為或轉換自成對的 FASTA 和 QUAL 檔案。
從 FASTQ 轉換為 FASTA 很簡單
from Bio import SeqIO
SeqIO.convert("example.fastq", "fastq", "example.fasta", "fasta")
從 FASTQ 轉換為 QUAL 也很簡單
from Bio import SeqIO
SeqIO.convert("example.fastq", "fastq", "example.qual", "qual")
但是,反向轉換會比較棘手。您可以使用 Bio.SeqIO.parse() 來逐一查看單一檔案中的記錄,但在這種情況下,我們有兩個輸入檔案。有幾種可能的策略,但假設兩個檔案確實成對,最節省記憶體的方式是一起迴圈處理。程式碼有點複雜,因此我們在 Bio.SeqIO.QualityIO 模組中提供一個名為 PairedFastaQualIterator 的函數來執行此操作。此函數接受兩個處理代碼(FASTA 檔案和 QUAL 檔案),並傳回 SeqRecord 迭代器
from Bio.SeqIO.QualityIO import PairedFastaQualIterator
for record in PairedFastaQualIterator(open("example.fasta"), open("example.qual")):
print(record)
此函數會檢查 FASTA 和 QUAL 檔案是否一致(例如,記錄是否以相同順序排列,且具有相同序列長度)。您可以將此函數與 Bio.SeqIO.write() 函數結合使用,將一對 FASTA 和 QUAL 檔案轉換為單一 FASTQ 檔案
from Bio import SeqIO
from Bio.SeqIO.QualityIO import PairedFastaQualIterator
with open("example.fasta") as f_handle, open("example.qual") as q_handle:
records = PairedFastaQualIterator(f_handle, q_handle)
count = SeqIO.write(records, "temp.fastq", "fastq")
print("Converted %i records" % count)
為 FASTQ 檔案建立索引
FASTQ 檔案通常非常大,其中包含數百萬筆讀取資料。由於資料量龐大,您無法一次將所有記錄載入記憶體。這就是為什麼上面的範例(篩選和修剪)會逐一查看檔案,一次只查看一個 SeqRecord。
但是,有時候您無法使用大型迴圈或迭代器 - 您可能需要隨機存取讀取資料。此時,Bio.SeqIO.index() 函數可能會非常有用,因為它允許您按名稱存取 FASTQ 檔案中的任何讀取資料(請參閱第 將序列檔案作為字典 – 索引檔案 節)。
我們將再次使用 ENA 的 SRR020192.fastq 檔案 (ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR020/SRR020192/SRR020192.fastq.gz),雖然這實際上是一個相當小的 FASTQ 檔案,讀取資料少於 \(50,000\) 筆
>>> from Bio import SeqIO
>>> fq_dict = SeqIO.index("SRR020192.fastq", "fastq")
>>> len(fq_dict)
41892
>>> list(fq_dict.keys())[:4]
['SRR020192.38240', 'SRR020192.23181', 'SRR020192.40568', 'SRR020192.23186']
>>> fq_dict["SRR020192.23186"].seq
Seq('GTCCCAGTATTCGGATTTGTCTGCCAAAACAATGAAATTGACACAGTTTACAAC...CCG')
在具有七百萬筆讀取資料的 FASTQ 檔案上測試時,建立索引大約需要一分鐘,但記錄存取幾乎是立即的。
姊妹函數 Bio.SeqIO.index_db() 可讓您將索引儲存到 SQLite3 資料庫檔案,以便幾乎立即重複使用 - 請參閱第 將序列檔案作為字典 – 索引檔案 節以取得更多詳細資訊。
第 排序序列檔案 節中的範例說明如何使用 Bio.SeqIO.index() 函數來排序大型 FASTA 檔案 – 這也可以用於 FASTQ 檔案。
轉換 SFF 檔案
如果您使用 454 (Roche) 序列資料,您可能可以存取標準流圖格式 (SFF) 檔案中的原始資料。這包含具有品質評分和原始流資訊的序列讀取資料(稱為鹼基)。
常見的任務是將 SFF 轉換為一對 FASTA 和 QUAL 檔案,或轉換為單一 FASTQ 檔案。使用 Bio.SeqIO.convert() 函數(請參閱第 在序列檔案格式之間轉換 節)即可輕鬆完成這些操作
>>> from Bio import SeqIO
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff", "reads.fasta", "fasta")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff", "reads.qual", "qual")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff", "reads.fastq", "fastq")
10
請記住,convert 函數會傳回記錄數量,在此範例中只有十個。這會為您提供未修剪的讀取資料,其中前導和尾隨品質不良的序列或接頭會以小寫字母表示。如果您想要修剪後的讀取資料(使用 SFF 檔案中記錄的剪輯資訊),請使用以下程式碼
>>> from Bio import SeqIO
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff-trim", "trimmed.fasta", "fasta")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff-trim", "trimmed.qual", "qual")
10
>>> SeqIO.convert("E3MFGYR02_random_10_reads.sff", "sff-trim", "trimmed.fastq", "fastq")
10
如果您執行 Linux,您可以向 Roche 要求其「儀器外」工具的副本(通常稱為 Newbler 工具)。這提供了一種替代方法,可在命令列執行 SFF 到 FASTA 或 QUAL 的轉換(但目前不支援 FASTQ 輸出),例如
$ sffinfo -seq -notrim E3MFGYR02_random_10_reads.sff > reads.fasta
$ sffinfo -qual -notrim E3MFGYR02_random_10_reads.sff > reads.qual
$ sffinfo -seq -trim E3MFGYR02_random_10_reads.sff > trimmed.fasta
$ sffinfo -qual -trim E3MFGYR02_random_10_reads.sff > trimmed.qual
Biopython 使用混合大小寫序列字串來表示修剪點的方式,刻意模仿 Roche 工具的做法。
如需 Biopython SFF 支援的更多資訊,請參閱內建說明
>>> from Bio.SeqIO import SffIO
>>> help(SffIO)
識別開放閱讀框架
識別可能基因的第一個非常簡單的步驟是尋找開放閱讀框架 (ORF)。我們指的是在所有六個框架中尋找沒有停止密碼子的長區域 – ORF 只是沒有框架內停止密碼子的核苷酸區域。
當然,若要尋找基因,您還需要擔心定位起始密碼子、可能的啟動子 – 以及在真核生物中,還有內含子需要擔心。但是,這種方法在病毒和原核生物中仍然很有用。
若要說明您如何使用 Biopython 來處理此問題,我們需要一個要搜尋的序列,而作為範例,我們將再次使用細菌質體 – 雖然這次我們將從沒有預先標記基因的純 FASTA 檔案開始:NC_005816.fna。這是一個細菌序列,因此我們將使用 NCBI 密碼子表 11(請參閱第 轉譯 節,瞭解轉譯)。
>>> from Bio import SeqIO
>>> record = SeqIO.read("NC_005816.fna", "fasta")
>>> table = 11
>>> min_pro_len = 100
以下是一個巧妙的技巧,使用 Seq 物件的 split 方法,以取得六個閱讀框架中所有可能的 ORF 轉譯的清單
>>> for strand, nuc in [(+1, record.seq), (-1, record.seq.reverse_complement())]:
... for frame in range(3):
... length = 3 * ((len(record) - frame) // 3) # Multiple of three
... for pro in nuc[frame : frame + length].translate(table).split("*"):
... if len(pro) >= min_pro_len:
... print(
... "%s...%s - length %i, strand %i, frame %i"
... % (pro[:30], pro[-3:], len(pro), strand, frame)
... )
...
GCLMKKSSIVATIITILSGSANAASSQLIP...YRF - length 315, strand 1, frame 0
KSGELRQTPPASSTLHLRLILQRSGVMMEL...NPE - length 285, strand 1, frame 1
GLNCSFFSICNWKFIDYINRLFQIIYLCKN...YYH - length 176, strand 1, frame 1
VKKILYIKALFLCTVIKLRRFIFSVNNMKF...DLP - length 165, strand 1, frame 1
NQIQGVICSPDSGEFMVTFETVMEIKILHK...GVA - length 355, strand 1, frame 2
RRKEHVSKKRRPQKRPRRRRFFHRLRPPDE...PTR - length 128, strand 1, frame 2
TGKQNSCQMSAIWQLRQNTATKTRQNRARI...AIK - length 100, strand 1, frame 2
QGSGYAFPHASILSGIAMSHFYFLVLHAVK...CSD - length 114, strand -1, frame 0
IYSTSEHTGEQVMRTLDEVIASRSPESQTR...FHV - length 111, strand -1, frame 0
WGKLQVIGLSMWMVLFSQRFDDWLNEQEDA...ESK - length 125, strand -1, frame 1
RGIFMSDTMVVNGSGGVPAFLFSGSTLSSY...LLK - length 361, strand -1, frame 1
WDVKTVTGVLHHPFHLTFSLCPEGATQSGR...VKR - length 111, strand -1, frame 1
LSHTVTDFTDQMAQVGLCQCVNVFLDEVTG...KAA - length 107, strand -1, frame 2
RALTGLSAPGIRSQTSCDRLRELRYVPVSL...PLQ - length 119, strand -1, frame 2
請注意,在此處,我們是從每個鏈的 5' 端 (起始) 開始計算框架。從正向鏈的 5' 端 (起始) 開始計算有時候會更容易。
您可以輕鬆編輯上述基於迴圈的程式碼,以建立候選蛋白質的清單,或將此程式碼轉換為清單理解。現在,此程式碼沒有做的一件事是追蹤蛋白質的位置。
您可以使用幾種方法來解決此問題。例如,下列程式碼會根據蛋白質計數來追蹤位置,並透過乘以三再針對框架和鏈調整來轉換回父序列
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
table = 11
min_pro_len = 100
def find_orfs_with_trans(seq, trans_table, min_protein_length):
answer = []
seq_len = len(seq)
for strand, nuc in [(+1, seq), (-1, seq.reverse_complement())]:
for frame in range(3):
trans = nuc[frame:].translate(trans_table)
trans_len = len(trans)
aa_start = 0
aa_end = 0
while aa_start < trans_len:
aa_end = trans.find("*", aa_start)
if aa_end == -1:
aa_end = trans_len
if aa_end - aa_start >= min_protein_length:
if strand == 1:
start = frame + aa_start * 3
end = min(seq_len, frame + aa_end * 3 + 3)
else:
start = seq_len - frame - aa_end * 3 - 3
end = seq_len - frame - aa_start * 3
answer.append((start, end, strand, trans[aa_start:aa_end]))
aa_start = aa_end + 1
answer.sort()
return answer
orf_list = find_orfs_with_trans(record.seq, table, min_pro_len)
for start, end, strand, pro in orf_list:
print(
"%s...%s - length %i, strand %i, %i:%i"
% (pro[:30], pro[-3:], len(pro), strand, start, end)
)
以及輸出
NQIQGVICSPDSGEFMVTFETVMEIKILHK...GVA - length 355, strand 1, 41:1109
WDVKTVTGVLHHPFHLTFSLCPEGATQSGR...VKR - length 111, strand -1, 491:827
KSGELRQTPPASSTLHLRLILQRSGVMMEL...NPE - length 285, strand 1, 1030:1888
RALTGLSAPGIRSQTSCDRLRELRYVPVSL...PLQ - length 119, strand -1, 2830:3190
RRKEHVSKKRRPQKRPRRRRFFHRLRPPDE...PTR - length 128, strand 1, 3470:3857
GLNCSFFSICNWKFIDYINRLFQIIYLCKN...YYH - length 176, strand 1, 4249:4780
RGIFMSDTMVVNGSGGVPAFLFSGSTLSSY...LLK - length 361, strand -1, 4814:5900
VKKILYIKALFLCTVIKLRRFIFSVNNMKF...DLP - length 165, strand 1, 5923:6421
LSHTVTDFTDQMAQVGLCQCVNVFLDEVTG...KAA - length 107, strand -1, 5974:6298
GCLMKKSSIVATIITILSGSANAASSQLIP...YRF - length 315, strand 1, 6654:7602
IYSTSEHTGEQVMRTLDEVIASRSPESQTR...FHV - length 111, strand -1, 7788:8124
WGKLQVIGLSMWMVLFSQRFDDWLNEQEDA...ESK - length 125, strand -1, 8087:8465
TGKQNSCQMSAIWQLRQNTATKTRQNRARI...AIK - length 100, strand 1, 8741:9044
QGSGYAFPHASILSGIAMSHFYFLVLHAVK...CSD - length 114, strand -1, 9264:9609
如果您註解掉排序陳述式,則蛋白質序列會以與之前相同的順序顯示,因此您可以檢查此程式碼是否正在執行相同的操作。在此處,我們已依位置將它們排序,以使其更容易與 GenBank 檔案中的實際註解進行比較(如第 一個不錯的範例 節中所視覺化的)。
但是,如果您只想找到開放閱讀框架的位置,則轉換每個可能的密碼子(包括進行反向互補以搜尋反向鏈)會浪費時間。您需要做的只是搜尋可能的停止密碼子(及其反向互補)。在此處使用正規表示式是顯而易見的方法(請參閱 Python 模組 re)。這些是描述搜尋字串的非常強大(但相當複雜)的方法,許多程式設計語言以及命令列工具(如 grep)都支援這些方法。您可以找到關於此主題的整本書籍!
序列剖析加上簡單繪圖
本節顯示一些序列剖析的範例,使用第 序列輸入/輸出 章中所述的 Bio.SeqIO 模組,以及 Python 程式庫 matplotlib 的 pylab 繪圖介面(請參閱 matplotlib 網站上的教學課程)。請注意,若要遵循這些範例,您需要安裝 matplotlib - 但即使沒有安裝 matplotlib,您仍然可以嘗試資料剖析部分。
序列長度直方圖
在許多時候,您可能會想要視覺化資料集中序列長度的分佈 – 例如,在基因體組裝專案中,疊圖的大小範圍。在此範例中,我們將重複使用我們的蘭花 FASTA 檔案 ls_orchid.fasta,其中只有 94 個序列。
首先,我們將使用 Bio.SeqIO 來剖析 FASTA 檔案,並編譯所有序列長度的清單。您可以使用 for 迴圈來執行此操作,但我發現清單理解更令人愉快
>>> from Bio import SeqIO
>>> sizes = [len(rec) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")]
>>> len(sizes), min(sizes), max(sizes)
(94, 572, 789)
>>> sizes
[740, 753, 748, 744, 733, 718, 730, 704, 740, 709, 700, 726, ..., 592]
現在我們有了所有基因的長度(以整數列表的形式),我們可以使用 matplotlib 的直方圖函數來顯示它。
from Bio import SeqIO
sizes = [len(rec) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")]
import pylab
pylab.hist(sizes, bins=20)
pylab.title(
"%i orchid sequences\nLengths %i to %i" % (len(sizes), min(sizes), max(sizes))
)
pylab.xlabel("Sequence length (bp)")
pylab.ylabel("Count")
pylab.show()
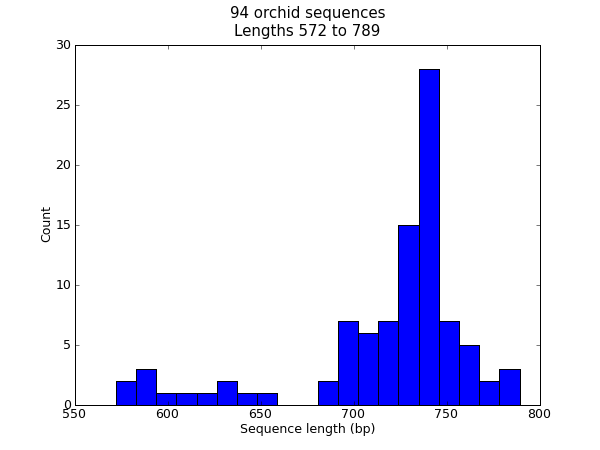
圖 22 蘭花序列長度的直方圖。
這應該會彈出一個新視窗,其中包含 圖 22 中顯示的圖形。請注意,大多數蘭花序列的長度約為 \(740\) 個鹼基對,而且這裡可能存在兩種不同的序列類型,其中一部分序列較短。
提示: 除了使用 pylab.show() 在視窗中顯示繪圖之外,您也可以使用 pylab.savefig(...) 將圖形儲存到檔案(例如,以 PNG 或 PDF 格式)。
序列 GC% 的繪圖
核苷酸序列另一個容易計算的量是 GC%。例如,您可能想查看細菌基因組中所有基因的 GC%,並研究任何可能最近透過水平基因轉移獲得的異常值。同樣,對於此範例,我們將重複使用我們的蘭花 FASTA 檔案 ls_orchid.fasta。
首先,我們將使用 Bio.SeqIO 來解析 FASTA 檔案並編譯所有 GC 百分比的列表。同樣,您可以使用 for 迴圈來完成此操作,但我更喜歡這個方式
from Bio import SeqIO
from Bio.SeqUtils import gc_fraction
gc_values = sorted(
100 * gc_fraction(rec.seq) for rec in SeqIO.parse("ls_orchid.fasta", "fasta")
)
讀取每個序列並計算 GC% 後,我們將它們按升序排序。現在,我們將使用 matplotlib 繪製這個浮點數列表
import pylab
pylab.plot(gc_values)
pylab.title(
"%i orchid sequences\nGC%% %0.1f to %0.1f"
% (len(gc_values), min(gc_values), max(gc_values))
)
pylab.xlabel("Genes")
pylab.ylabel("GC%")
pylab.show()
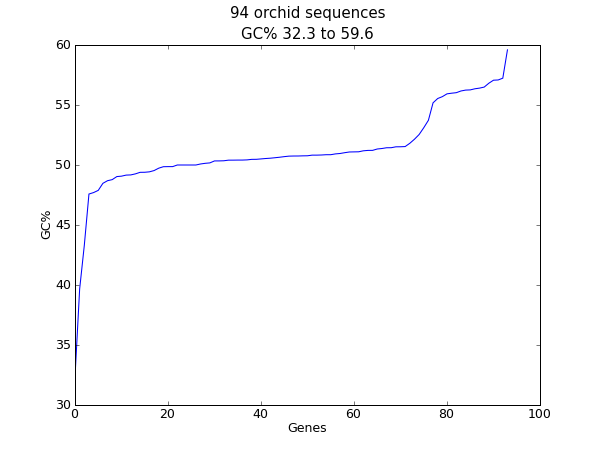
圖 23 蘭花序列長度的直方圖。
與先前的範例一樣,這應該會彈出一個新視窗,其中包含 圖 23 中顯示的圖形。如果您嘗試在一個生物體的完整基因組上執行此操作,您可能會得到比這平滑得多的圖形。
核苷酸點圖
點圖是一種視覺比較兩個核苷酸序列彼此相似性的方法。使用滑動視窗來比較短的子序列彼此,通常會設定一個不匹配閾值。為了簡單起見,這裡我們只尋找完全匹配(在 圖 24 中以黑色顯示)。
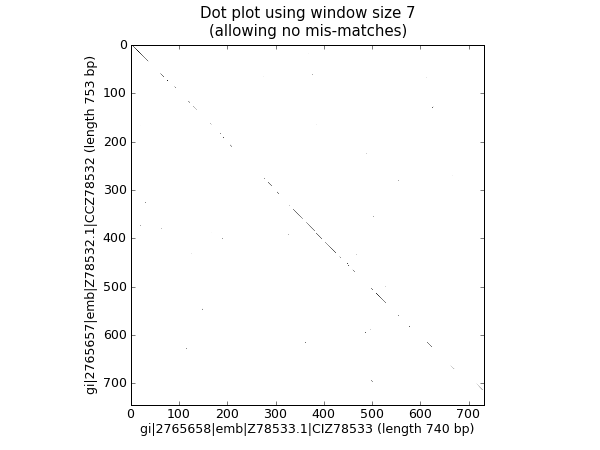
圖 24 使用圖像顯示的兩個蘭花序列的核苷酸點圖。
首先,我們需要兩個序列。為了方便起見,我們將從我們的蘭花 FASTA 檔案 ls_orchid.fasta 中取出前兩個序列
from Bio import SeqIO
with open("ls_orchid.fasta") as in_handle:
record_iterator = SeqIO.parse(in_handle, "fasta")
rec_one = next(record_iterator)
rec_two = next(record_iterator)
我們將展示兩種方法。首先,一個簡單的樸素實作,它將所有視窗大小的子序列相互比較,以編譯相似度矩陣。您可以建構一個矩陣或陣列物件,但這裡我們只使用一個以巢狀列表理解建立的布林值列表
window = 7
seq_one = rec_one.seq.upper()
seq_two = rec_two.seq.upper()
data = [
[
(seq_one[i : i + window] != seq_two[j : j + window])
for j in range(len(seq_one) - window)
]
for i in range(len(seq_two) - window)
]
請注意,我們在這裡沒有檢查反向互補匹配。現在,我們將使用 matplotlib 的 pylab.imshow() 函數來顯示此數據,首先請求灰色配色方案,以便以黑白顯示
import pylab
pylab.gray()
pylab.imshow(data)
pylab.xlabel("%s (length %i bp)" % (rec_one.id, len(rec_one)))
pylab.ylabel("%s (length %i bp)" % (rec_two.id, len(rec_two)))
pylab.title("Dot plot using window size %i\n(allowing no mis-matches)" % window)
pylab.show()
這應該會彈出一個新視窗,顯示 圖 24 中的圖形。正如您可能預期的那樣,這兩個序列非常相似,沿著對角線有一條部分視窗大小的匹配線。沒有非對角線匹配,這表示存在倒位或其他有趣的事件。
上面的程式碼在小型範例上可以正常運作,但是將其應用於較大的序列時會有兩個問題,我們將在下面解決。首先,這種針對所有比較的暴力方法非常慢。相反,我們將編譯將視窗大小的子序列對應到其位置的字典,然後取集合交集以找出兩個序列中都找到的那些子序列。這會使用更多記憶體,但速度快得多。其次,pylab.imshow() 函數可以顯示的矩陣大小有限。作為替代方案,我們將使用 pylab.scatter() 函數。
我們首先建立將視窗大小的子序列對應到位置的字典
window = 7
dict_one = {}
dict_two = {}
for seq, section_dict in [
(rec_one.seq.upper(), dict_one),
(rec_two.seq.upper(), dict_two),
]:
for i in range(len(seq) - window):
section = seq[i : i + window]
try:
section_dict[section].append(i)
except KeyError:
section_dict[section] = [i]
# Now find any sub-sequences found in both sequences
matches = set(dict_one).intersection(dict_two)
print("%i unique matches" % len(matches))
為了使用 pylab.scatter(),我們需要單獨的列表來表示 \(x\) 和 \(y\) 座標
# Create lists of x and y coordinates for scatter plot
x = []
y = []
for section in matches:
for i in dict_one[section]:
for j in dict_two[section]:
x.append(i)
y.append(j)
我們現在準備好將修改後的點圖繪製為散佈圖
import pylab
pylab.cla() # clear any prior graph
pylab.gray()
pylab.scatter(x, y)
pylab.xlim(0, len(rec_one) - window)
pylab.ylim(0, len(rec_two) - window)
pylab.xlabel("%s (length %i bp)" % (rec_one.id, len(rec_one)))
pylab.ylabel("%s (length %i bp)" % (rec_two.id, len(rec_two)))
pylab.title("Dot plot using window size %i\n(allowing no mis-matches)" % window)
pylab.show()
這應該會彈出一個新視窗,顯示 圖 25 中的圖形。
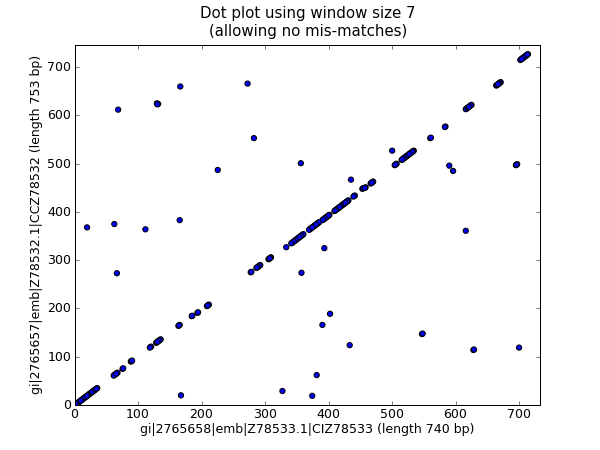
圖 25 使用散佈圖的兩個蘭花序列的核苷酸點圖。
我個人覺得第二個圖更容易閱讀!再次請注意,我們在這裡沒有檢查反向互補匹配 – 您可以擴展此範例來執行此操作,並且或許可以用一種顏色繪製正向匹配,用另一種顏色繪製反向匹配。
繪製定序讀取數據的品質分數
如果您正在處理第二代定序數據,您可能想嘗試繪製品質數據。以下是一個使用兩個包含配對末端讀取的 FASTQ 檔案的範例,SRR001666_1.fastq 用於正向讀取,而 SRR001666_2.fastq 用於反向讀取。這些是從 ENA 定序讀取資料庫 FTP 網站下載的(ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR001/SRR001666/SRR001666_1.fastq.gz 和 ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR001/SRR001666/SRR001666_2.fastq.gz),並且來自大腸桿菌 – 有關詳細資訊,請參閱 https://www.ebi.ac.uk/ena/data/view/SRR001666。
在下面的程式碼中,使用 pylab.subplot(...) 函數來並排顯示兩個子圖上的正向和反向品質。還有一些程式碼只繪製前 50 個讀取。
import pylab
from Bio import SeqIO
for subfigure in [1, 2]:
filename = "SRR001666_%i.fastq" % subfigure
pylab.subplot(1, 2, subfigure)
for i, record in enumerate(SeqIO.parse(filename, "fastq")):
if i >= 50:
break # trick!
pylab.plot(record.letter_annotations["phred_quality"])
pylab.ylim(0, 45)
pylab.ylabel("PHRED quality score")
pylab.xlabel("Position")
pylab.savefig("SRR001666.png")
print("Done")
您應該注意,我們在這裡使用 Bio.SeqIO 格式名稱 fastq,因為 NCBI 使用帶有 PHRED 分數的標準 Sanger FASTQ 格式儲存了這些讀取。但是,正如您從讀取長度中猜測的那樣,此數據來自 Illumina Genome Analyzer,並且最初可能採用兩種 Solexa/Illumina FASTQ 變體檔案格式之一。
此範例使用 pylab.savefig(...) 函數代替 pylab.show(...),但如前所述,兩者都很有用。
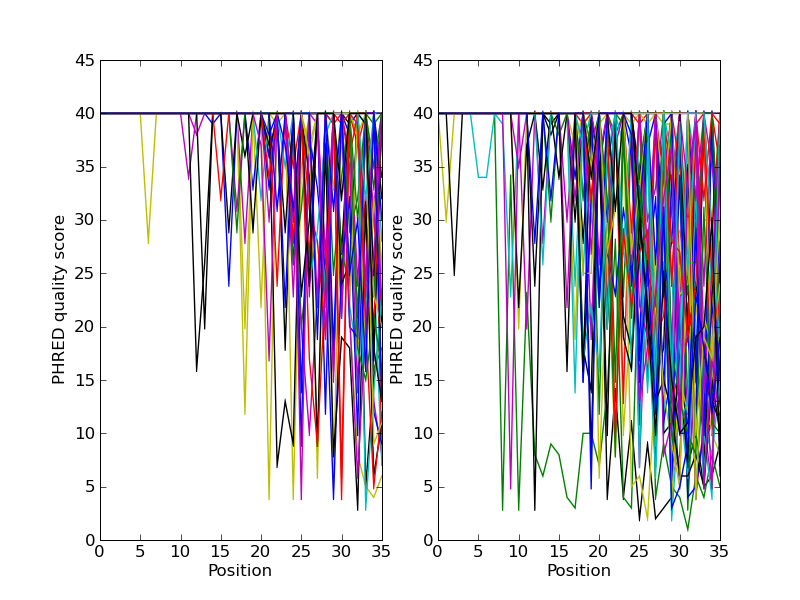
圖 26 一些配對末端讀取的品質圖。
結果顯示在 圖 26 中。
BioSQL – 將序列儲存在關聯式資料庫中
BioSQL 是 OBF 專案(BioPerl、BioJava 等)之間的共同努力,旨在支援用於儲存序列資料的共享資料庫綱要。理論上,您可以使用 BioPerl 將 GenBank 檔案載入到資料庫中,然後使用 Biopython 從資料庫中將其提取為帶有功能的記錄物件 - 並獲得或多或少與直接使用 Bio.SeqIO (第 序列輸入/輸出 章節) 將 GenBank 檔案載入為 SeqRecord 相同的結果。
Biopython 的 BioSQL 模組目前記錄在 https://biopython.dev.org.tw/wiki/BioSQL,這是我們維基頁面的一部分。