圖形包含 GenomeDiagram
Bio.Graphics 模組依賴於第三方 Python 函式庫 ReportLab。雖然 ReportLab 主要用於產生 PDF 檔案,但它也可以建立封裝式 PostScript (EPS) 和 (SVG) 檔案。除了這些向量式影像之外,如果安裝了某些其他依賴項(例如 Python Imaging Library (PIL)),ReportLab 也可以輸出點陣圖影像(包括 JPEG、PNG、GIF、BMP 和 PICT 格式)。
GenomeDiagram
簡介
Bio.Graphics.GenomeDiagram 模組已新增至 Biopython 1.50,之前是作為依賴 Biopython 的獨立 Python 模組提供。GenomeDiagram 在 Pritchard 等人 (2006) 的《生物資訊學》期刊出版物中有所描述 [Pritchard2006],其中包含一些範例圖片。舊版手冊的 PDF 副本在此處 https://biopython.dev.org.tw/DIST/docs/GenomeDiagram/userguide.pdf,其中包含更多範例。
顧名思義,GenomeDiagram 旨在繪製整個基因組,特別是原核基因組,以線性圖(可選擇分割成片段以更好地符合)或環狀輪圖的形式。請參閱 Toth *等人* (2006) 中的圖 2 [Toth2006],以獲得良好的範例。它也適用於為較小的基因組(如噬菌體、質體或粒線體)繪製相當詳細的圖,例如,參閱 Van der Auwera *等人* (2009) 中的圖 1 和圖 2 [Vanderauwera2009](顯示為額外的手動編輯)。
如果您已將基因組載入為包含許多 SeqFeature 物件的 SeqRecord 物件(例如從 GenBank 檔案載入的),則此模組最容易使用(請參閱章節 序列註解物件 和 序列輸入/輸出)。
圖表、軌道、特徵集和特徵
GenomeDiagram 使用一組巢狀物件。在最上層,您有一個圖表物件,代表沿著水平軸(或圓形)的序列(或序列區域)。一個圖表可以包含一個或多個軌道,這些軌道以垂直方式堆疊(或在圓形圖表上以放射狀排列)。這些軌道通常都具有相同的長度,並且代表相同的序列區域。您可以使用一個軌道來顯示基因位置、另一個軌道來顯示調控區域,以及第三個軌道來顯示 GC 百分比。
最常用的軌道類型將包含特徵,這些特徵捆綁在特徵集中。您可以選擇為所有 CDS 特徵使用一個特徵集,而為 tRNA 特徵使用另一個特徵集。這不是必需的,它們都可以放在同一個特徵集中,但這樣可以更輕鬆地更新僅選定特徵的屬性(例如,將所有 tRNA 特徵設為紅色)。
有兩種主要方式可以建立完整的圖表。首先,由上而下的方法,您建立一個圖表物件,然後使用其方法新增軌道,並使用軌道方法新增特徵集,並使用其方法新增特徵。其次,您可以單獨建立個別物件(以適合您程式碼的任何順序),然後將它們組合起來。
由上而下的範例
我們將從 GenBank 檔案讀取的 SeqRecord 物件中繪製整個基因組(請參閱章節 序列輸入/輸出)。此範例使用來自 *鼠疫耶爾辛氏菌微小變種* 的 pPCP1 質體,該檔案包含在 Biopython 單元測試的 GenBank 資料夾下,或在線上 NC_005816.gb 從我們的網站。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
我們使用由上而下的方法,因此在載入我們的序列之後,我們接下來建立一個空的圖表,然後新增一個(空的)軌道,並將一個(空的)特徵集新增到其中
gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()
現在是有趣的部分 - 我們取得 SeqRecord 中的每個基因 SeqFeature 物件,並使用它在圖表上產生一個特徵。我們將它們塗成藍色,在深藍色和淺藍色之間交替。
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, color=color, label=True)
現在我們來實際製作輸出檔案。這分兩個步驟進行,首先我們呼叫 draw 方法,該方法使用 ReportLab 物件建立所有形狀。然後,我們呼叫 write 方法,該方法將這些形狀呈現為要求的檔案格式。請注意,您可以輸出多種檔案格式
gd_diagram.draw(
format="linear",
orientation="landscape",
pagesize="A4",
fragments=4,
start=0,
end=len(record),
)
gd_diagram.write("plasmid_linear.pdf", "PDF")
gd_diagram.write("plasmid_linear.eps", "EPS")
gd_diagram.write("plasmid_linear.svg", "SVG")
此外,如果您已安裝依賴項,您也可以執行點陣圖,例如
gd_diagram.write("plasmid_linear.png", "PNG")
預期輸出顯示在 圖 9 中。
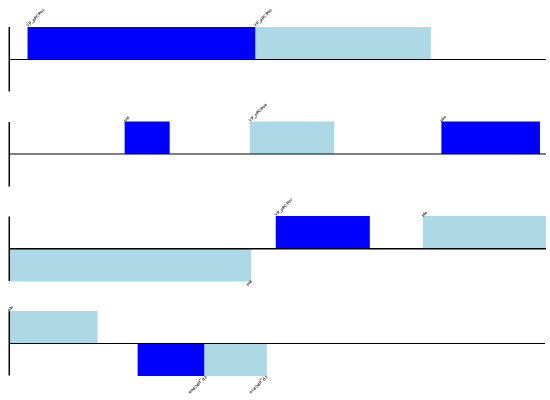
圖 9 *鼠疫耶爾辛氏菌微小變種* 質體 pPCP1 的簡單線性圖。
請注意,我們設定為四的 fragments 引數會控制基因組被分割成多少個片段。
如果您想要製作圓形圖,請嘗試此方法
gd_diagram.draw(
format="circular",
circular=True,
pagesize=(20 * cm, 20 * cm),
start=0,
end=len(record),
circle_core=0.7,
)
gd_diagram.write("plasmid_circular.pdf", "PDF")
預期輸出顯示在 圖 10 中。
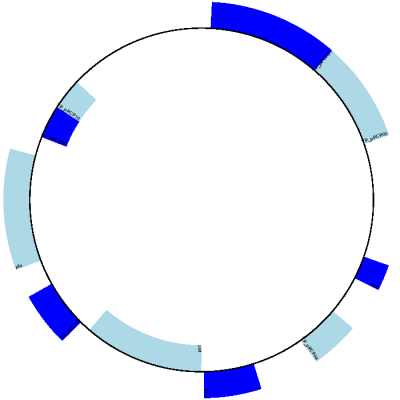
圖 10 *鼠疫耶爾辛氏菌微小變種* 質體 pPCP1 的簡單圓形圖。
這些圖形不是很令人興奮,但我們才剛開始。
由下而上的範例
現在讓我們使用由下而上的方法產生完全相同的圖形。這表示我們直接建立不同的物件(而且幾乎可以以任何順序執行),然後將它們組合起來。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
record = SeqIO.read("NC_005816.gb", "genbank")
# Create the feature set and its feature objects,
gd_feature_set = GenomeDiagram.FeatureSet()
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(feature, color=color, label=True)
# (this for loop is the same as in the previous example)
# Create a track, and a diagram
gd_track_for_features = GenomeDiagram.Track(name="Annotated Features")
gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
# Now have to glue the bits together...
gd_track_for_features.add_set(gd_feature_set)
gd_diagram.add_track(gd_track_for_features, 1)
您現在可以像之前一樣呼叫 draw 和 write 方法,以使用上方由上而下範例末尾的程式碼產生線性或圓形圖。這些圖形應該是相同的。
沒有 SeqFeature 的特徵
在上述範例中,我們使用 SeqRecord 的 SeqFeature 物件來建立我們的圖表(另請參閱章節 特徵、位置和方位物件)。有時您不會有 SeqFeature 物件,而只有要繪製的特徵的座標。您必須建立最小的 SeqFeature 物件,但這很容易
from Bio.SeqFeature import SeqFeature, SimpleLocation
my_seq_feature = SeqFeature(SimpleLocation(50, 100, strand=+1))
對於鏈,請使用 +1 表示正向鏈,-1 表示反向鏈,以及 None 表示兩者。以下是一個簡短的自我包含範例
from Bio.SeqFeature import SeqFeature, SimpleLocation
from Bio.Graphics import GenomeDiagram
from reportlab.lib.units import cm
gdd = GenomeDiagram.Diagram("Test Diagram")
gdt_features = gdd.new_track(1, greytrack=False)
gds_features = gdt_features.new_set()
# Add three features to show the strand options,
feature = SeqFeature(SimpleLocation(25, 125, strand=+1))
gds_features.add_feature(feature, name="Forward", label=True)
feature = SeqFeature(SimpleLocation(150, 250, strand=None))
gds_features.add_feature(feature, name="Strandless", label=True)
feature = SeqFeature(SimpleLocation(275, 375, strand=-1))
gds_features.add_feature(feature, name="Reverse", label=True)
gdd.draw(format="linear", pagesize=(15 * cm, 4 * cm), fragments=1, start=0, end=400)
gdd.write("GD_labels_default.pdf", "pdf")
輸出顯示在 圖 11 的頂部(以預設特徵顏色淺綠色顯示)。
請注意,我們在這裡使用了 name 引數來指定這些特徵的標題文字。接下來將更詳細地討論此內容。
特徵標題
回想一下,我們使用以下程式碼(其中 feature 是 SeqFeature 物件)將特徵新增至圖表
gd_feature_set.add_feature(feature, color=color, label=True)
在上述範例中,SeqFeature 註解用於為特徵選擇合理的標題。預設情況下,使用 SeqFeature 物件的限定詞字典下的以下可能條目:gene、label、name、locus_tag 和 product。更簡單地說,您可以直接指定名稱
gd_feature_set.add_feature(feature, color=color, label=True, name="My Gene")
除了每個特徵標籤的標題文字之外,您還可以選擇字型、位置(預設為符號的開頭,您也可以選擇中間或結尾)和方向(僅適用於線性圖,其中預設旋轉 \(45\) 度)
# Large font, parallel with the track
gd_feature_set.add_feature(
feature, label=True, color="green", label_size=25, label_angle=0
)
# Very small font, perpendicular to the track (towards it)
gd_feature_set.add_feature(
feature,
label=True,
color="purple",
label_position="end",
label_size=4,
label_angle=90,
)
# Small font, perpendicular to the track (away from it)
gd_feature_set.add_feature(
feature,
label=True,
color="blue",
label_position="middle",
label_size=6,
label_angle=-90,
)
將這三個片段的每一個與上一節中的完整範例組合在一起,應該會產生類似 圖 11 中的軌道。
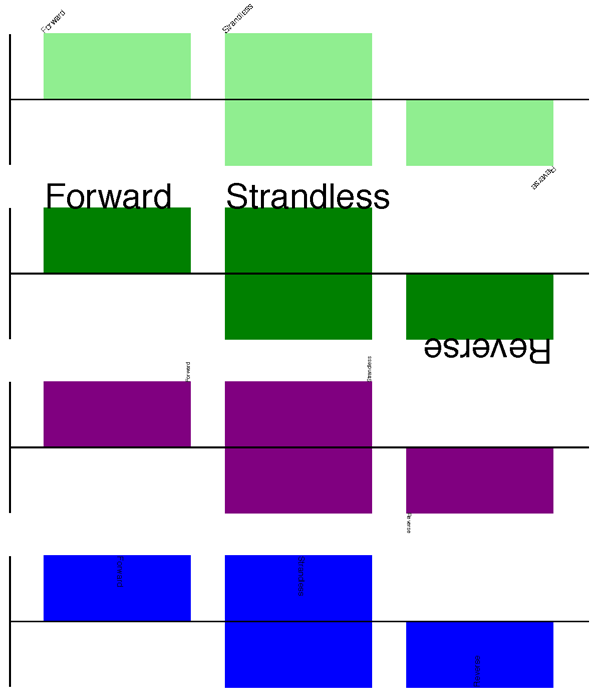
圖 11 簡單的 GenomeDiagram 展示標籤選項。
淺綠色的頂部繪圖顯示預設標籤設定(請參閱沒有 SeqFeature 的特徵章節),而其餘部分則顯示標籤大小、位置和方向的變化(請參閱特徵標題章節)。
我們這裡沒有展示,但您也可以設定 label_color 來控制標籤的顏色(在一個很好的範例章節中使用)。
您會注意到預設字體非常小 - 這是合理的,因為您通常會在頁面上繪製許多(小)特徵,而不僅僅是這裡顯示的幾個大特徵。
特徵符號
上面的範例都只使用了特徵的預設符號,一個普通的方塊,這是在 GenomeDiagram 最後公開發布的獨立版本中唯一可用的。當 GenomeDiagram 加入 Biopython 1.50 時,包含了箭頭符號。
# Default uses a BOX sigil
gd_feature_set.add_feature(feature)
# You can make this explicit:
gd_feature_set.add_feature(feature, sigil="BOX")
# Or opt for an arrow:
gd_feature_set.add_feature(feature, sigil="ARROW")
Biopython 1.61 添加了三個符號,
# Box with corners cut off (making it an octagon)
gd_feature_set.add_feature(feature, sigil="OCTO")
# Box with jagged edges (useful for showing breaks in contains)
gd_feature_set.add_feature(feature, sigil="JAGGY")
# Arrow which spans the axis with strand used only for direction
gd_feature_set.add_feature(feature, sigil="BIGARROW")
這些顯示在圖 12 中。大多數符號都符合一個邊界框(如預設的 BOX 符號所示),在正向或反向鏈的軸之上或之下,或跨在軸上(高度加倍),適用於無鏈特徵。BIGARROW 符號不同,總是跨在軸上,方向取決於特徵的鏈。

圖 12 簡單的 GenomeDiagram 展示不同的符號。
箭頭符號
我們在上一節中介紹了箭頭符號。有兩個額外的選項可以調整箭頭的形狀,首先是箭頭軸的粗細,以邊界框高度的比例給出
# Full height shafts, giving pointed boxes:
gd_feature_set.add_feature(feature, sigil="ARROW", color="brown", arrowshaft_height=1.0)
# Or, thin shafts:
gd_feature_set.add_feature(feature, sigil="ARROW", color="teal", arrowshaft_height=0.2)
# Or, very thin shafts:
gd_feature_set.add_feature(
feature, sigil="ARROW", color="darkgreen", arrowshaft_height=0.1
)
結果顯示在圖 13中。
圖 13 簡單的 GenomeDiagram 展示箭頭軸選項。
其次,箭頭頭部的長度 - 以邊界框高度的比例給出(預設為 \(0.5\),或 \(50\%\))
# Short arrow heads:
gd_feature_set.add_feature(feature, sigil="ARROW", color="blue", arrowhead_length=0.25)
# Or, longer arrow heads:
gd_feature_set.add_feature(feature, sigil="ARROW", color="orange", arrowhead_length=1)
# Or, very very long arrow heads (i.e. all head, no shaft, so triangles):
gd_feature_set.add_feature(feature, sigil="ARROW", color="red", arrowhead_length=10000)
結果顯示在圖 14中。
圖 14 簡單的 GenomeDiagram 展示箭頭頭部選項。
Biopython 1.61 添加了一個新的 BIGARROW 符號,它總是跨在軸上,反向鏈指向左邊,否則指向右邊
# A large arrow straddling the axis:
gd_feature_set.add_feature(feature, sigil="BIGARROW")
上面顯示的 ARROW 符號的所有軸和箭頭頭部選項也可用於 BIGARROW 符號。
一個很好的範例
現在讓我們回到來自鼠疫耶爾森氏菌微型變種的 pPCP1 質體,以及由上而下的範例章節中使用的由上而下的方法,但利用我們現在討論過的符號選項。這次我們將對基因使用箭頭,並將它們與顯示一些限制酶切位點位置的無鏈特徵(作為普通方塊)疊加在一起。
from reportlab.lib import colors
from reportlab.lib.units import cm
from Bio.Graphics import GenomeDiagram
from Bio import SeqIO
from Bio.SeqFeature import SeqFeature, SimpleLocation
record = SeqIO.read("NC_005816.gb", "genbank")
gd_diagram = GenomeDiagram.Diagram(record.id)
gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
gd_feature_set = gd_track_for_features.new_set()
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
if len(gd_feature_set) % 2 == 0:
color = colors.blue
else:
color = colors.lightblue
gd_feature_set.add_feature(
feature, sigil="ARROW", color=color, label=True, label_size=14, label_angle=0
)
# I want to include some strandless features, so for an example
# will use EcoRI recognition sites etc.
for site, name, color in [
("GAATTC", "EcoRI", colors.green),
("CCCGGG", "SmaI", colors.orange),
("AAGCTT", "HindIII", colors.red),
("GGATCC", "BamHI", colors.purple),
]:
index = 0
while True:
index = record.seq.find(site, start=index)
if index == -1:
break
feature = SeqFeature(SimpleLocation(index, index + len(site)))
gd_feature_set.add_feature(
feature,
color=color,
name=name,
label=True,
label_size=10,
label_color=color,
)
index += len(site)
gd_diagram.draw(format="linear", pagesize="A4", fragments=4, start=0, end=len(record))
gd_diagram.write("plasmid_linear_nice.pdf", "PDF")
gd_diagram.write("plasmid_linear_nice.eps", "EPS")
gd_diagram.write("plasmid_linear_nice.svg", "SVG")
gd_diagram.draw(
format="circular",
circular=True,
pagesize=(20 * cm, 20 * cm),
start=0,
end=len(record),
circle_core=0.5,
)
gd_diagram.write("plasmid_circular_nice.pdf", "PDF")
gd_diagram.write("plasmid_circular_nice.eps", "EPS")
gd_diagram.write("plasmid_circular_nice.svg", "SVG")
預期的輸出顯示在顯示所選限制酶切位點的質體 pPCP1 線性圖。和顯示所選限制酶切位點的質體 pPCP1 環狀圖。。
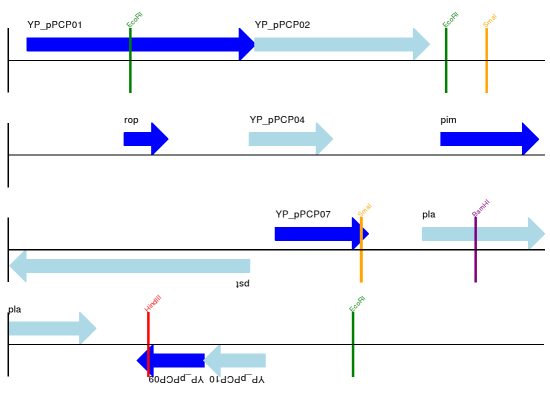
圖 15 顯示所選限制酶切位點的質體 pPCP1 線性圖。
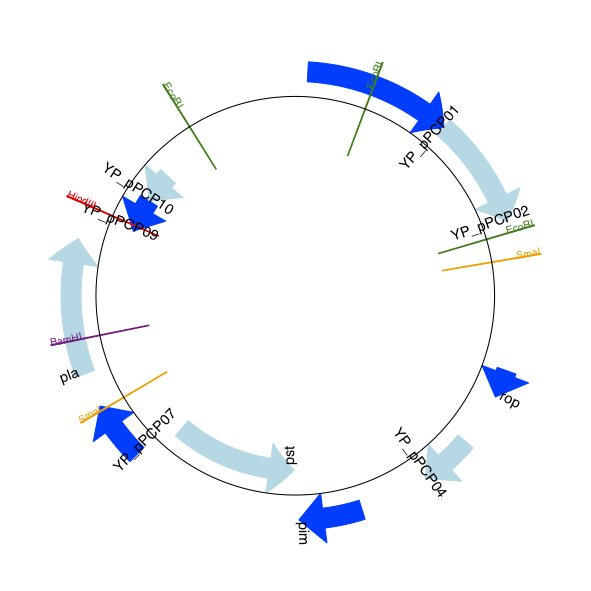
圖 16 顯示所選限制酶切位點的質體 pPCP1 環狀圖。
多個軌道
到目前為止,所有範例都使用單個軌道,但您可以有多個軌道 – 例如,在一個軌道上顯示基因,在另一個軌道上顯示重複區域。在這個範例中,我們將並排顯示三個噬菌體基因組,並按比例縮放,靈感來自 Proux et al. (2002) [Proux2002] 的圖 6。我們將需要以下三個噬菌體的 GenBank 檔案
NC_002703– 乳酸桿菌噬菌體 Tuc2009,完整基因組 (\(38347\) bp)AF323668– 噬菌體 bIL285,完整基因組 (\(35538\) bp)NC_003212– 無害李斯特菌 Clip11262,完整基因組,我們只關注其中的整合前噬菌體 5(長度相似)。
如果您願意,可以使用 Entrez 下載這些,請參閱EFetch:從 Entrez 下載完整記錄章節以了解更多詳細資訊。對於第三個記錄,我們已經計算出噬菌體整合到基因組中的位置,並將記錄切片以提取它(保留特徵,請參閱切片 SeqRecord章節),並且還必須反向互補以匹配前兩個噬菌體的方向(再次保留特徵,請參閱反向互補 SeqRecord 物件章節)
from Bio import SeqIO
A_rec = SeqIO.read("NC_002703.gbk", "gb")
B_rec = SeqIO.read("AF323668.gbk", "gb")
C_rec = SeqIO.read("NC_003212.gbk", "gb")[2587879:2625807].reverse_complement(name=True)
我們模仿的圖使用了不同的顏色來表示不同的基因功能。一種方法是編輯 GenBank 檔案以記錄每個特徵的顏色偏好 - 桑格的 Artemis 編輯器 確實可以做到這一點,而 GenomeDiagram 應該可以理解。然而,在這裡,我們只會硬編碼三個顏色列表。
請注意,GenBank 檔案中的註釋與 Proux et al. 中顯示的註釋並不完全匹配,他們繪製了一些未註釋的基因。
from reportlab.lib.colors import (
red,
grey,
orange,
green,
brown,
blue,
lightblue,
purple,
)
A_colors = (
[red] * 5
+ [grey] * 7
+ [orange] * 2
+ [grey] * 2
+ [orange]
+ [grey] * 11
+ [green] * 4
+ [grey]
+ [green] * 2
+ [grey, green]
+ [brown] * 5
+ [blue] * 4
+ [lightblue] * 5
+ [grey, lightblue]
+ [purple] * 2
+ [grey]
)
B_colors = (
[red] * 6
+ [grey] * 8
+ [orange] * 2
+ [grey]
+ [orange]
+ [grey] * 21
+ [green] * 5
+ [grey]
+ [brown] * 4
+ [blue] * 3
+ [lightblue] * 3
+ [grey] * 5
+ [purple] * 2
)
C_colors = (
[grey] * 30
+ [green] * 5
+ [brown] * 4
+ [blue] * 2
+ [grey, blue]
+ [lightblue] * 2
+ [grey] * 5
)
現在來繪製它們 – 這次我們在圖表中添加三個軌道,並且注意到它們被賦予不同的起始/結束值以反映它們的不同長度(這需要 Biopython 1.59 或更高版本)。
from Bio.Graphics import GenomeDiagram
name = "Proux Fig 6"
gd_diagram = GenomeDiagram.Diagram(name)
max_len = 0
for record, gene_colors in zip([A_rec, B_rec, C_rec], [A_colors, B_colors, C_colors]):
max_len = max(max_len, len(record))
gd_track_for_features = gd_diagram.new_track(
1, name=record.name, greytrack=True, start=0, end=len(record)
)
gd_feature_set = gd_track_for_features.new_set()
i = 0
for feature in record.features:
if feature.type != "gene":
# Exclude this feature
continue
gd_feature_set.add_feature(
feature,
sigil="ARROW",
color=gene_colors[i],
label=True,
name=str(i + 1),
label_position="start",
label_size=6,
label_angle=0,
)
i += 1
gd_diagram.draw(format="linear", pagesize="A4", fragments=1, start=0, end=max_len)
gd_diagram.write(name + ".pdf", "PDF")
gd_diagram.write(name + ".eps", "EPS")
gd_diagram.write(name + ".svg", "SVG")
預期的輸出顯示在圖 17中。
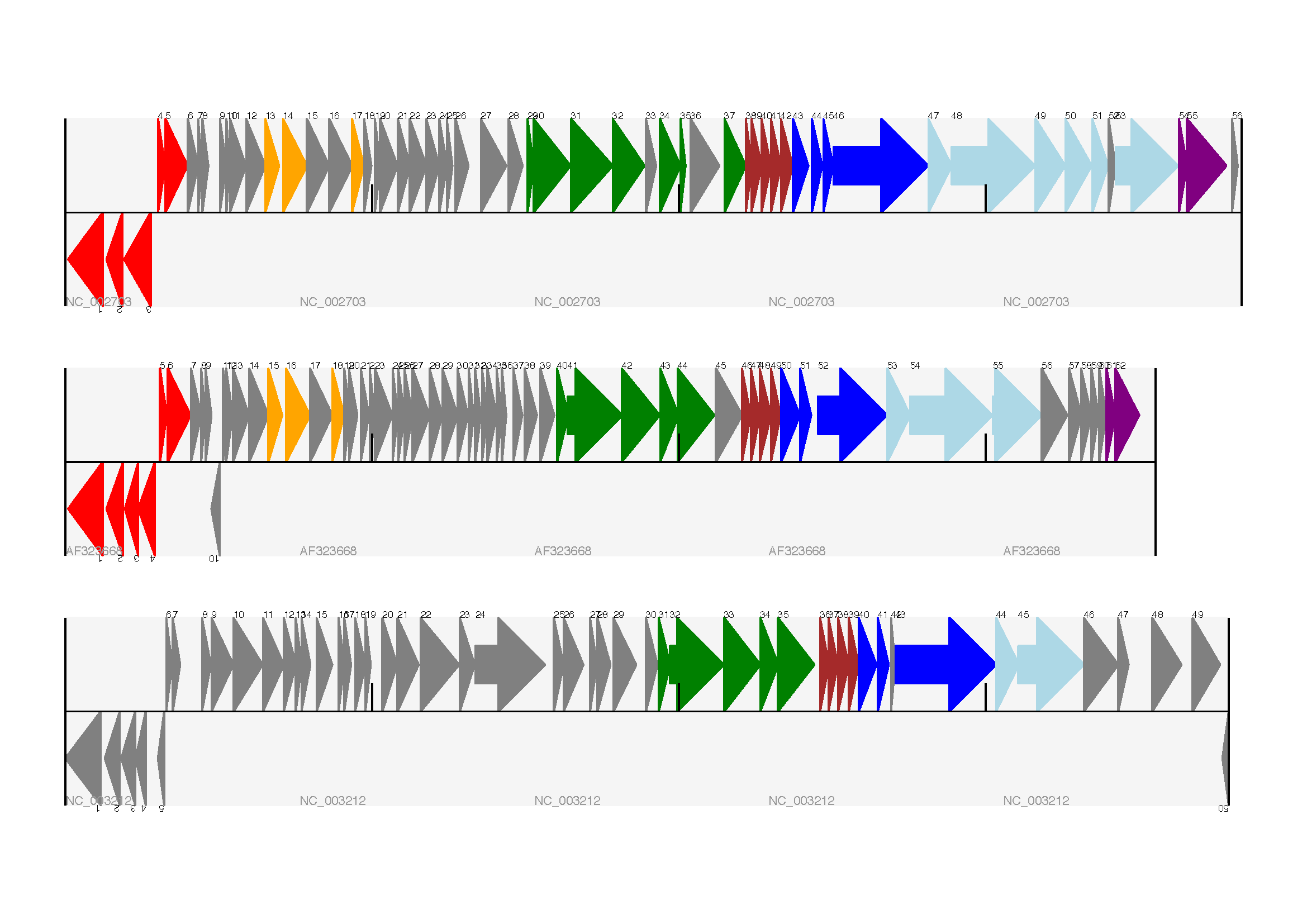
圖 17 三個噬菌體的三個軌道的線性圖。
這顯示了乳酸桿菌噬菌體 Tuc2009 (NC_002703)、噬菌體 bIL285 (AF323668) 和來自無害李斯特菌 Clip11262 (NC_003212) 的前噬菌體 5(請參閱多個軌道章節)。
我確實想知道為什麼在原始手稿中,底部噬菌體中沒有標記紅色或橙色基因。另一個重點是,這裡顯示的噬菌體長度不同 - 這是因為它們都是按相同比例繪製的(它們確實長度不同)。
與已發布圖表的關鍵差異在於,它們在相似的蛋白質之間具有顏色編碼的連結 – 這就是我們在下一節中所要做的。
軌道之間的交叉連結
Biopython 1.59 添加了在軌道之間繪製交叉連結的功能 - 這裡將展示簡單的線性圖,以及分割成片段的線性圖和環狀圖。
繼續前一節中靈感來自 Proux et al. 2002 [Proux2002] 的圖 6 的範例,我們將需要基因對之間交叉連結的列表,以及要使用的分數或顏色。實際上,您可能會從 BLAST 檔案中計算得出此結果,但這裡我已手動輸入它們。
我的命名約定繼續將這三個噬菌體稱為 A、B 和 C。以下是我們要顯示的 A 和 B 之間的連結,以元組列表形式給出(相似性分數百分比、A 中的基因、B 中的基因)。
# Tuc2009 (NC_002703) vs bIL285 (AF323668)
A_vs_B = [
(99, "Tuc2009_01", "int"),
(33, "Tuc2009_03", "orf4"),
(94, "Tuc2009_05", "orf6"),
(100, "Tuc2009_06", "orf7"),
(97, "Tuc2009_07", "orf8"),
(98, "Tuc2009_08", "orf9"),
(98, "Tuc2009_09", "orf10"),
(100, "Tuc2009_10", "orf12"),
(100, "Tuc2009_11", "orf13"),
(94, "Tuc2009_12", "orf14"),
(87, "Tuc2009_13", "orf15"),
(94, "Tuc2009_14", "orf16"),
(94, "Tuc2009_15", "orf17"),
(88, "Tuc2009_17", "rusA"),
(91, "Tuc2009_18", "orf20"),
(93, "Tuc2009_19", "orf22"),
(71, "Tuc2009_20", "orf23"),
(51, "Tuc2009_22", "orf27"),
(97, "Tuc2009_23", "orf28"),
(88, "Tuc2009_24", "orf29"),
(26, "Tuc2009_26", "orf38"),
(19, "Tuc2009_46", "orf52"),
(77, "Tuc2009_48", "orf54"),
(91, "Tuc2009_49", "orf55"),
(95, "Tuc2009_52", "orf60"),
]
同樣適用於 B 和 C
# bIL285 (AF323668) vs Listeria innocua prophage 5 (in NC_003212)
B_vs_C = [
(42, "orf39", "lin2581"),
(31, "orf40", "lin2580"),
(49, "orf41", "lin2579"), # terL
(54, "orf42", "lin2578"), # portal
(55, "orf43", "lin2577"), # protease
(33, "orf44", "lin2576"), # mhp
(51, "orf46", "lin2575"),
(33, "orf47", "lin2574"),
(40, "orf48", "lin2573"),
(25, "orf49", "lin2572"),
(50, "orf50", "lin2571"),
(48, "orf51", "lin2570"),
(24, "orf52", "lin2568"),
(30, "orf53", "lin2567"),
(28, "orf54", "lin2566"),
]
對於第一個和最後一個噬菌體,這些識別符是 locus 標籤,對於中間的噬菌體,沒有 locus 標籤,所以我使用了基因名稱代替。以下的小型輔助函數可讓我們使用 locus 標籤或基因名稱來查找特徵
def get_feature(features, id, tags=["locus_tag", "gene"]):
"""Search list of SeqFeature objects for an identifier under the given tags."""
for f in features:
for key in tags:
# tag may not be present in this feature
for x in f.qualifiers.get(key, []):
if x == id:
return f
raise KeyError(id)
我們現在可以將這些識別符對列表轉換為 SeqFeature 對,從而找到它們的位置座標。我們現在可以將所有程式碼和以下程式碼片段新增到先前的範例(就在 gd_diagram.draw(...) 行之前 – 請參閱Proux_et_al_2002_Figure_6.py 包含在 Biopython 原始碼的 Doc/examples 資料夾中)以將交叉連結新增到圖表中
from Bio.Graphics.GenomeDiagram import CrossLink
from reportlab.lib import colors
# Note it might have been clearer to assign the track numbers explicitly...
for rec_X, tn_X, rec_Y, tn_Y, X_vs_Y in [
(A_rec, 3, B_rec, 2, A_vs_B),
(B_rec, 2, C_rec, 1, B_vs_C),
]:
track_X = gd_diagram.tracks[tn_X]
track_Y = gd_diagram.tracks[tn_Y]
for score, id_X, id_Y in X_vs_Y:
feature_X = get_feature(rec_X.features, id_X)
feature_Y = get_feature(rec_Y.features, id_Y)
color = colors.linearlyInterpolatedColor(
colors.white, colors.firebrick, 0, 100, score
)
link_xy = CrossLink(
(track_X, feature_X.location.start, feature_X.location.end),
(track_Y, feature_Y.location.start, feature_Y.location.end),
color,
colors.lightgrey,
)
gd_diagram.cross_track_links.append(link_xy)
此程式碼中有幾個重要的部分。首先,GenomeDiagram 物件有一個 cross_track_links 屬性,它只是一個 CrossLink 物件的列表。每個 CrossLink 物件都採用兩組特定於軌道的座標(這裡以元組形式給出,您也可以使用 GenomeDiagram.Feature 物件代替)。您可以選擇性地提供顏色、邊框顏色,並說明是否應翻轉繪製此連結(對於顯示反轉很有用)。
您還可以瞭解我們如何將 BLAST 百分比識別分數轉換為顏色,在白色 (\(0\%\)) 和深紅色 (\(100\%\)) 之間插值。在這個範例中,我們沒有任何重疊的交叉連結問題。解決這個問題的一種方法是在 ReportLab 中使用透明度,方法是使用設定了 alpha 通道的顏色。但是,這種著色的顏色方案與重疊透明度組合在一起會難以解釋。預期的輸出顯示在圖 18中。
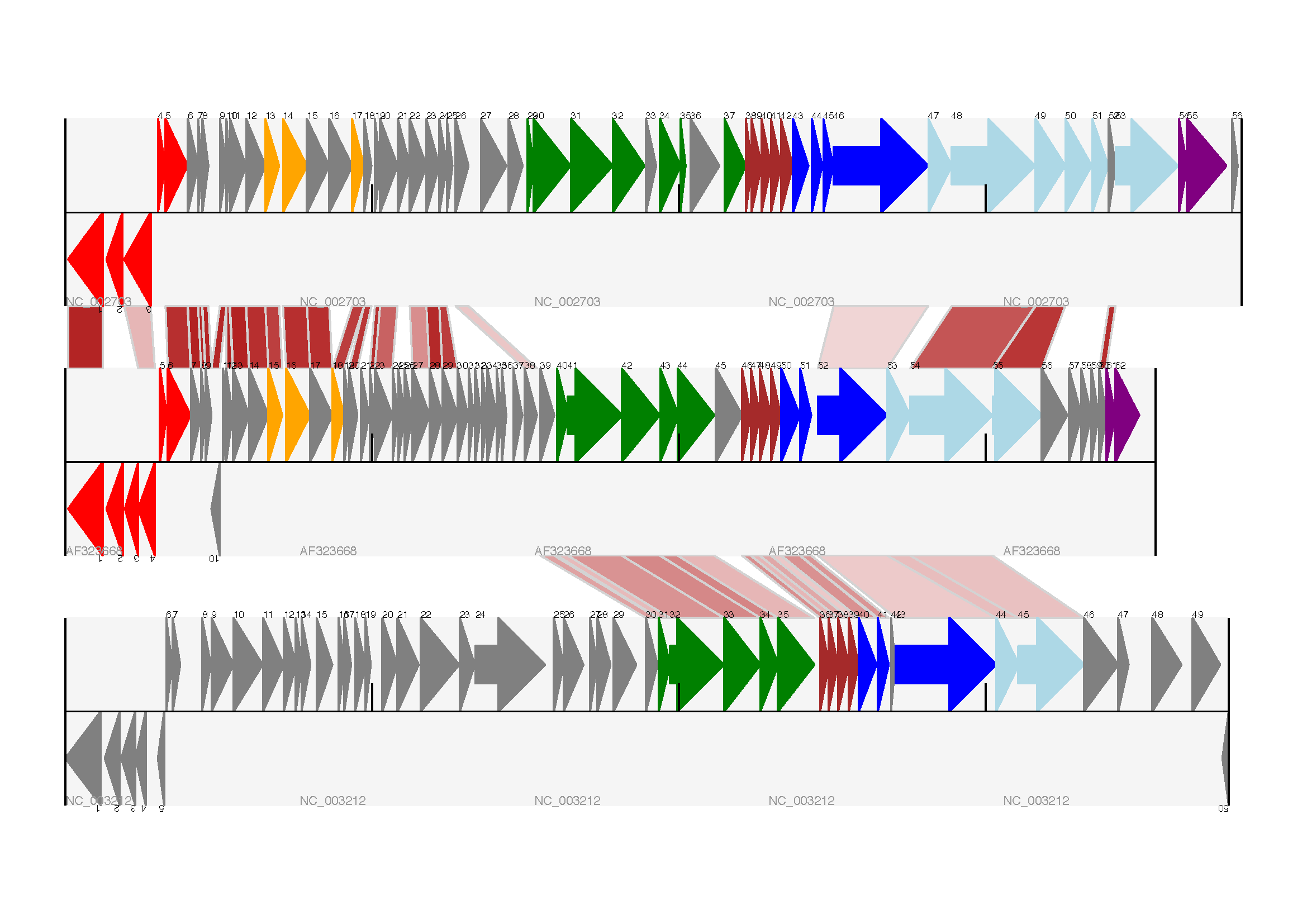
圖 18 具有三個軌道加上基本交叉連結的線性圖。
這三個軌道顯示了乳酸球菌噬菌體 Tuc2009 (NC_002703)、噬菌體 bIL285 (AF323668),以及來自單核細胞增生李斯特菌 Clip11262 的前噬菌體 5 (NC_003212),並以百分比相似度著色的基本交叉連結(請參閱軌道間的交叉連結章節)。
在 Biopython 中,還有很多可以改進此圖表的地方。首先,這裡的交叉連結位於以鏈特異性方式繪製的蛋白質之間。在特徵軌道上添加背景區域(使用「BOX」符號的特徵)以延伸交叉連結可能會有所幫助。此外,我們可以減少特徵軌道的垂直高度,以便為連結分配更多空間 - 一種方法是為空軌道分配空間。此外,在像這種沒有大型基因重疊的情況下,我們可以使用跨軸的 BIGARROW 符號,這可以讓我們進一步減少軌道所需的垂直空間。這些改進在範例腳本 Proux_et_al_2002_Figure_6.py 中展示,該腳本包含在 Biopython 原始碼的 Doc/examples 資料夾中。預期輸出結果顯示在圖 19中。
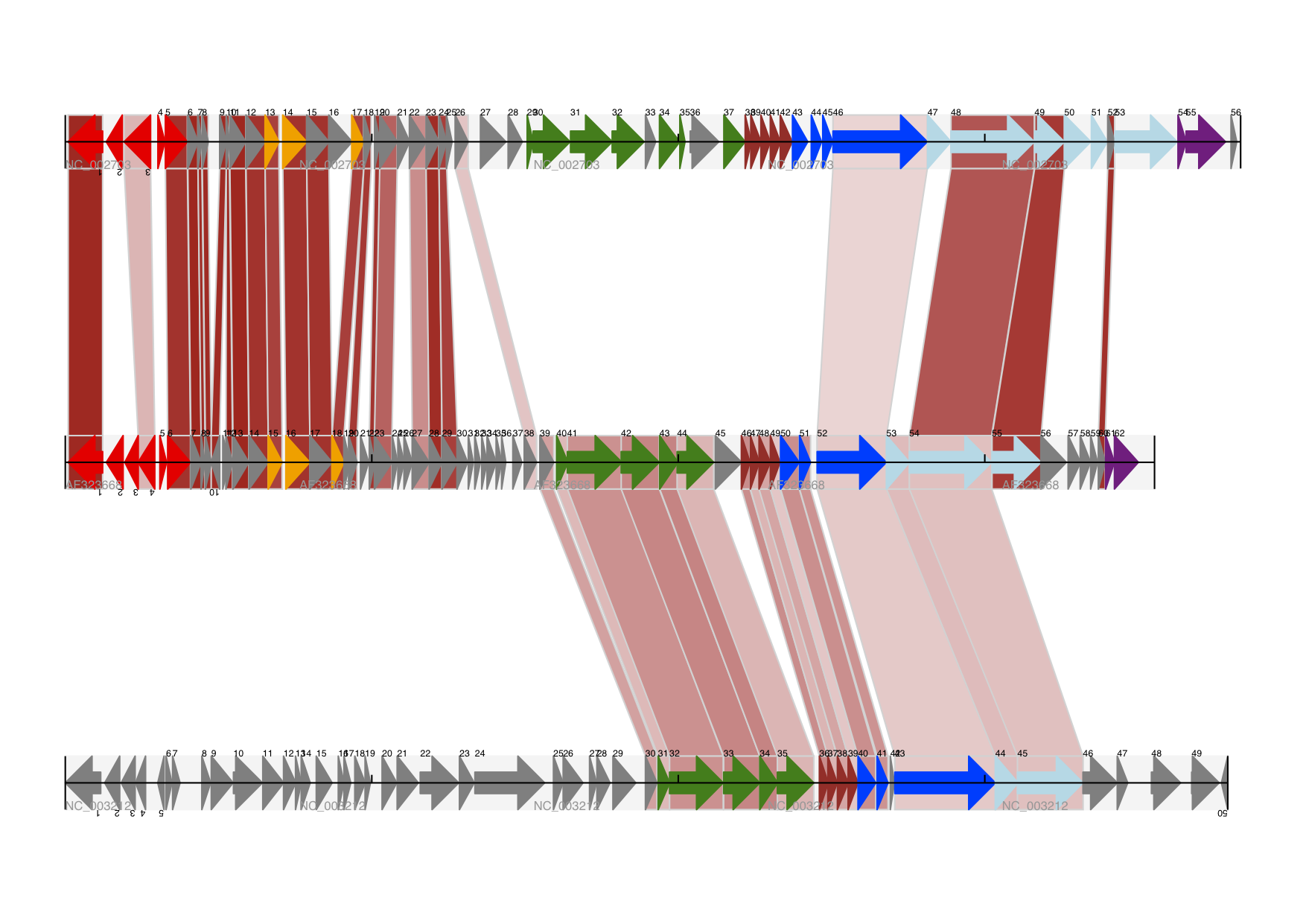
圖 19 具有三個軌道加上陰影交叉連結的線性圖。
這三個軌道顯示了乳酸球菌噬菌體 Tuc2009 (NC_002703)、噬菌體 bIL285 (AF323668),以及來自單核細胞增生李斯特菌 Clip11262 的前噬菌體 5 (NC_003212),以及按百分比相似度著色的交叉連結(請參閱軌道間的交叉連結章節)。
除此之外,您可能想要在向量影像編輯器中手動進行的潤飾包括微調基因標籤的位置,以及添加其他自訂註釋,例如突出顯示特定區域。
雖然在本例中並非真正必要,因為沒有任何交叉連結重疊,但在 ReportLab 中使用透明顏色是疊加多個連結的非常實用技巧。但是,在這種情況下,應避免使用陰影顏色方案。
更多選項
您可以控制刻度標記來顯示比例尺 - 畢竟,每個圖表都應顯示其單位以及灰色軌道標籤的數量。
此外,到目前為止我們只使用了 FeatureSet。GenomeDiagram 還有一個 GraphSet,可用於顯示折線圖、長條圖和熱圖(例如,顯示與特徵平行的軌道上的 GC% 圖)。
這些選項目前尚未涵蓋,因此現在我們將您推薦到 GenomeDiagram 獨立版本中包含的使用者指南 (PDF)(但請先閱讀下一節)和 docstrings。
轉換舊程式碼
如果您有使用 GenomeDiagram 獨立版本編寫的舊程式碼,並且想要將其切換為使用 Biopython 中包含的新版本,則必須進行一些更改 - 最重要的是對您的 import 語句進行更改。
此外,舊版本的 GenomeDiagram 僅使用 color 和 center 的英國拼法(colour 和 centre)。您需要更改為美國拼法,儘管幾年來 Biopython 版本的 GenomeDiagram 都支援兩者。
例如,如果您過去有
from GenomeDiagram import GDFeatureSet, GDDiagram
gdd = GDDiagram("An example")
...
您可以像這樣切換 import 語句
from Bio.Graphics.GenomeDiagram import FeatureSet as GDFeatureSet, Diagram as GDDiagram
gdd = GDDiagram("An example")
...
希望這就足夠了。從長遠來看,您可能想要切換到新的名稱,但您必須更改更多的程式碼
from Bio.Graphics.GenomeDiagram import FeatureSet, Diagram
gdd = Diagram("An example")
...
或
from Bio.Graphics import GenomeDiagram
gdd = GenomeDiagram.Diagram("An example")
...
如果您遇到困難,請在 Biopython 郵件列表中尋求建議。一個問題是我們尚未包含舊模組 GenomeDiagram.GDUtilities。這包括一些與 GC% 相關的函數,稍後可能會合併到 Bio.SeqUtils 下。
染色體
Bio.Graphics.BasicChromosome 模組允許繪製染色體。在 Jupe et al. (2012) [Jupe2012](開放存取)中有一個範例使用顏色來突出顯示不同的基因家族。
簡單染色體
這裡有一個非常簡單的範例 - 我們將使用擬南芥。
圖 20 擬南芥的簡單染色體圖。
您可以跳過這部分,但我首先從 NCBI 的 FTP 站點 ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Arabidopsis_thaliana/ 下載了五個已排序的染色體作為五個獨立的 FASTA 檔案,然後使用 Bio.SeqIO 解析它們以找出它們的長度。您可以使用 GenBank 檔案執行此操作(並且下一個範例使用這些檔案來繪製特徵),但是如果只需要長度,則使用整個染色體的 FASTA 檔案會更快
from Bio import SeqIO
entries = [
("Chr I", "CHR_I/NC_003070.fna"),
("Chr II", "CHR_II/NC_003071.fna"),
("Chr III", "CHR_III/NC_003074.fna"),
("Chr IV", "CHR_IV/NC_003075.fna"),
("Chr V", "CHR_V/NC_003076.fna"),
]
for name, filename in entries:
record = SeqIO.read(filename, "fasta")
print(name, len(record))
這給出了五個染色體的長度,我們現在將在以下 BasicChromosome 模組的簡短演示中使用這些長度
from reportlab.lib.units import cm
from Bio.Graphics import BasicChromosome
entries = [
("Chr I", 30432563),
("Chr II", 19705359),
("Chr III", 23470805),
("Chr IV", 18585042),
("Chr V", 26992728),
]
max_len = 30432563 # Could compute this from the entries dict
telomere_length = 1000000 # For illustration
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7 * cm, 21 * cm) # A4 landscape
for name, length in entries:
cur_chromosome = BasicChromosome.Chromosome(name)
# Set the scale to the MAXIMUM length plus the two telomeres in bp,
# want the same scale used on all five chromosomes so they can be
# compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
# Add a body - using bp as the scale length here.
body = BasicChromosome.ChromosomeSegment()
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("simple_chrom.pdf", "Arabidopsis thaliana")
這應該會建立一個非常簡單的 PDF 檔案,如圖 20所示。此範例刻意簡短而精要。下一個範例顯示感興趣的特徵的位置。
帶註釋的染色體
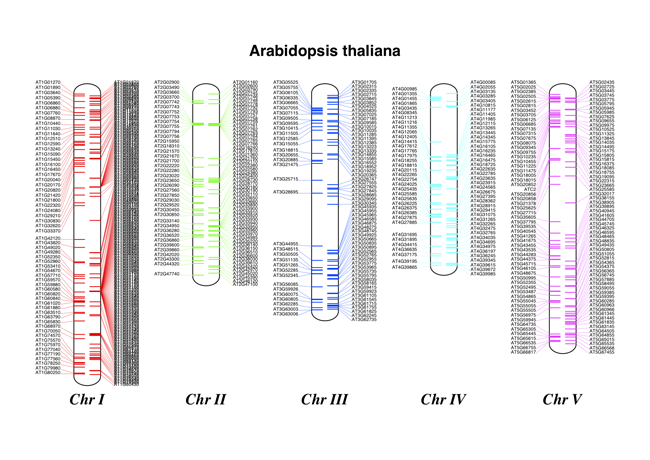
圖 21 顯示 tRNA 基因的擬南芥染色體圖。
從上一個範例繼續,我們也來顯示 tRNA 基因。我們將透過解析五個擬南芥染色體的 GenBank 檔案來取得它們的位置。您需要從 NCBI FTP 站點 ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Arabidopsis_thaliana/ 下載這些檔案,並保留子目錄名稱或編輯下方的路徑
from reportlab.lib.units import cm
from Bio import SeqIO
from Bio.Graphics import BasicChromosome
entries = [
("Chr I", "CHR_I/NC_003070.gbk"),
("Chr II", "CHR_II/NC_003071.gbk"),
("Chr III", "CHR_III/NC_003074.gbk"),
("Chr IV", "CHR_IV/NC_003075.gbk"),
("Chr V", "CHR_V/NC_003076.gbk"),
]
max_len = 30432563 # Could compute this from the entries dict
telomere_length = 1000000 # For illustration
chr_diagram = BasicChromosome.Organism()
chr_diagram.page_size = (29.7 * cm, 21 * cm) # A4 landscape
for index, (name, filename) in enumerate(entries):
record = SeqIO.read(filename, "genbank")
length = len(record)
features = [f for f in record.features if f.type == "tRNA"]
# Record an Artemis style integer color in the feature's qualifiers,
# 1 = Black, 2 = Red, 3 = Green, 4 = blue, 5 =cyan, 6 = purple
for f in features:
f.qualifiers["color"] = [index + 2]
cur_chromosome = BasicChromosome.Chromosome(name)
# Set the scale to the MAXIMUM length plus the two telomeres in bp,
# want the same scale used on all five chromosomes so they can be
# compared to each other
cur_chromosome.scale_num = max_len + 2 * telomere_length
# Add an opening telomere
start = BasicChromosome.TelomereSegment()
start.scale = telomere_length
cur_chromosome.add(start)
# Add a body - again using bp as the scale length here.
body = BasicChromosome.AnnotatedChromosomeSegment(length, features)
body.scale = length
cur_chromosome.add(body)
# Add a closing telomere
end = BasicChromosome.TelomereSegment(inverted=True)
end.scale = telomere_length
cur_chromosome.add(end)
# This chromosome is done
chr_diagram.add(cur_chromosome)
chr_diagram.draw("tRNA_chrom.pdf", "Arabidopsis thaliana")
它可能會警告您標籤太靠近 - 請查看 Chr I 的正向鏈(右側),但它應該會建立一個彩色 PDF 檔案,如圖 21所示。